
Submitted:
23 October 2025
Posted:
24 October 2025
You are already at the latest version
Abstract
The endocannabinoid system (eCBS) is a versatile neuromodulatory network that orchestrates synaptic plasticity, reward processing, and neuronal homeostasis. Increasing evidence implicates eCBS dysregulation in both addiction and neurodegenerative (ND) disorders, suggesting overlapping molecular and cellular mechanisms that underlie these conditions. This review synthesizes current advances in understanding how eCBS components—cannabinoid receptors (CB1 and CB2), endogenous ligands (anandamide and 2-arachidonoylglycerol), and their metabolic enzymes—modulate dopaminergic and glutamatergic signaling within reward and reinforcement circuits. Chronic exposure to drugs of abuse, including alcohol, cocaine, and methamphetamine, perturbs eCBS homeostasis, promoting oxidative stress, neuroinflammation, excitotoxicity, and protein aggregation—pathological features common to Alzheimer’s disease, Parkinson’s disease, Huntington’s disease, and amyotrophic lateral sclerosis. Experimental studies demonstrate that pharmacological or genetic manipulation of eCBS components can mitigate neurotoxic outcomes, emphasizing their homeostatic and neuroprotective potential. Despite these advances, mechanistic gaps remain regarding how substance-induced eCBS alterations precipitate neurodegenerative cascades. Addressing these gaps will be critical for leveraging the eCBS as a therapeutic target to counteract addiction-driven neurotoxicity and age-related cognitive decline.
Keywords:
cannabinoid receptors
; alcohol
; cell death
; brain circuits
; drugs of abuse
; drug reinforcement
; pathology
1. Introduction
The endocannabinoid system (eCBS) is a multifaceted signaling network composed of endocannabinoids (eCBs)—lipid-derived messengers synthesized from long-chain polyunsaturated fatty acids—along with their cognate cannabinoid receptors and the enzymes that regulate their biosynthesis and degradation. Together, these components orchestrate a wide range of physiological and pathological processes across the central nervous system (CNS) and peripheral tissues [1,2]. The system was initially characterized as the neuronal substrate through which the principal psychoactive constituent of cannabis, Δ9-tetrahydrocannabinol (Δ9-THC), exerts its behavioral and physiological effects. The first breakthrough occurred with the pharmacological characterization of the cannabinoid receptor type 1 (CB1) in the rat brain by Devane and colleagues [3,4,5], followed by the cloning of the gene encoding this G protein–coupled receptor (GPCR) by Matsuda and collaborators [6,7]. These landmark discoveries laid the foundation for deciphering the molecular basis of cannabis-induced psychoactivity. These discoveries unfolded the way for defining the broader physiological roles of the eCBS [2,8,9,10,11,12,13]. Shortly thereafter, a second receptor, cannabinoid receptor type 2 (CB2), was identified in the rat spleen [14].
CB1 is widely expressed throughout the CNS and peripheral organs [35,36,37]. In the brain, CB1 receptors are particularly enriched in the hippocampus, prefrontal cortex (PFC), basal ganglia, and cerebellum, with more moderate expression in the nucleus accumbens (NAc), thalamus, periaqueductal gray, and amygdala. Two major neuronal populations contribute to this distribution: gamma-aminobutyric acid (GABAergic) interneurons, which express high levels of CB1, and glutamatergic neurons, which express lower levels [38,39]. Additionally, CB1 receptors are present in monoaminergic nuclei such as the locus coeruleus (noradrenaline) and dorsal raphe nucleus (serotonin) [40,41], as well as in non-neuronal brain cells, including astrocytes, microglia, and oligodendrocytes [42,43]. Outside the CNS, CB1 receptors are expressed in the liver, gastrointestinal tract, skeletal muscle, reproductive tissues, adipocytes, immune cells, vascular tissues, and the cardiovascular system [44,45]. Activated CB1 receptors primarily couple to Gi/o proteins, leading to the inhibition of adenylyl cyclase and a reduction in intracellular cyclic adenosine monophosphate (cAMP) levels. Downstream signaling includes either activation [46,47,48,49,50,51,52] or inhibition [53,54,55] of mitogen-activated protein kinases (MAPKs) such as extracellular signal-regulated kinases (ERK), Jun N-terminal kinase (JNK), and p38, as well as modulation of cAMP-response element binding protein (CREB) phosphorylation, depending on cellular context (Figure 1). CB1 signaling also influences the phosphatidylinositol 3-kinase/ protein kinase B (PI3K/AKT) pathway, again in a context-dependent manner [53,54,56]. In neurons, CB1 activation suppresses voltage-gated calcium channels and enhances potassium channel conductance, thereby reducing intracellular Ca2+ and neurotransmitter release [51,57,58]. The released Gβγ subunits contribute to this effect by inhibiting N- and P/Q-type Ca2+ channels and activating A-type potassium channels [59,60]. In parallel, CB1-mediated Gβγ subunits initiate Src (non-receptor tyrosine kinase)-mediated cascades that activate MAPKs and focal adhesion kinases (FAK) [47,61].
Initially considered restricted to immune cells and peripheral tissues [35,62,63], CB2 receptors are now recognized as being expressed at low levels in the healthy CNS, particularly in microglia and astrocytes, with upregulation under pathological conditions such as brain injury, stroke, and neurodegeneration [64,65,66,67]. More recent studies have detected CB2 expression in neurons of the hippocampus, striatum, and thalamus [68,69], as well as dopaminergic neurons in the ventral tegmental area (VTA) [70]. Like CB1, CB2 couples predominantly to Gi/o proteins, inhibiting adenylyl cyclase and reducing cAMP levels (Figure 2) [35,71,72,73]. CB2 also activates the MAPK cascade, increasing ERK1/2 phosphorylation [62,72,74], and engages the PI3K–AKT pathway [75]. In addition, CB2 signaling can involve β-arrestin recruitment, which in some cases occurs independently of G protein-coupled receptor kinase (GRK) phosphorylation [76,77]. Ion channel modulation by CB2 includes inhibition of voltage-gated calcium channels (VGCC) and activation of GIRKs [78,79]. For detailed summaries of CB2 signaling, see recent reviews [80,81].
The cloning and characterization of CB1 also led to the identification of the first endogenous ligand, anandamide (AEA, N-arachidonoylethanolamine) (Figure 3) by the Mechoulam group in 1992 [5]. AEA only partially reproduced the psychotropic effects of Δ9-THC, leading to the subsequent discovery of a second major ligand, 2-arachidonoylglycerol (2-AG) [92,93] (Figure 3). Pharmacologically, AEA acts as a high-affinity partial agonist at CB1 with minimal CB2 activity, whereas 2-AG is a full agonist at both CB1 and CB2 but with lower binding affinity [94,95]. These distinctions highlight their complementary roles in regulating endocannabinoid tone. Beyond CB1/CB2, both AEA and 2-AG engage additional targets, including G-protein-coupled receptor 55 (GPR55) [95], transient receptor potential (TRP) channels such as TRPV1 [96], and nuclear receptors such as peroxisome proliferator-activated receptor alpha (PPARα) and gamma (PPARγ) [97], underscoring the pleiotropic nature of eCB signaling. Unlike classical neurotransmitters, eCBs are not stored in vesicles but are synthesized “on demand” in response to neuronal activity. Increases in intracellular calcium trigger their production and release from postsynaptic neurons, where they act as retrograde messengers at presynaptic CB1 receptors [98,99,100]. This mechanism modulates neurotransmitter release and can induce either short-term or long-term synaptic depression, depending on the circuit context [101]. AEA and 2-AG biosynthesis originates from membrane phospholipids. AEA is generated from N-acyl-phosphatidylethanolamine (NAPE) via NAPE-specific phospholipase D (NAPE-PLD), whereas 2-AG arises from diacylglycerol (DAG) through diacylglycerol lipase (DAGLα/β). Alternative enzymatic pathways for both ligands have also been described, adding further regulatory diversity [102,103]. Degradation is primarily hydrolytic: AEA by fatty acid amide hydrolase (FAAH), yielding arachidonic acid and ethanolamine, and 2-AG by monoacylglycerol lipase (MAGL), producing arachidonic acid and glycerol [104,105]. Both eCBs can also undergo oxidative metabolism via cyclooxygenase-2 (COX-2) and lipoxygenases, resulting in the generation of bioactive metabolites with distinct signaling properties [106]. The broad distribution of the eCBS across neural circuits and peripheral tissues underpins its involvement in diverse physiological processes, including cognition and memory, appetite regulation, motor control, pain modulation, immune responses, thermoregulation, sleep, stress adaptation, and reward processing [107]. In this review, we focus on recent advances in understanding the role of the eCBS in addiction and neurodegeneration, with particular emphasis on its dual significance as both a mediator of pathophysiology and a promising therapeutic target.
2. The Overview of the Reward and Reinforcement Circuit
The transition from recreational drug use to addiction represents a fundamental neurobiological shift in which voluntary consumption evolves into compulsive drug-seeking and taking, despite explicit awareness of harmful consequences. Progressive disruptions across multiple brain circuits drive this loss of control. Dysregulation within cortico-striatal and cortico-limbic networks alters reward sensitivity, incentive salience attribution, and associative conditioning, while impairments in prefrontal cortical regions compromise executive control, self-monitoring, and decision-making. In parallel, maladaptive changes in circuits regulating mood and interoceptive awareness exacerbate craving and heighten vulnerability to relapse. Collectively, these neuroadaptations provide a framework for understanding how chronic drug exposure reshapes brain function and entrenches the pathological state of addiction.
A central mechanism underlying this process is the mesolimbic dopamine system, which is consistently engaged by all well-studied drugs of abuse, including Δ9-THC. Classic studies demonstrated that such drugs elevate dopamine concentrations in terminal regions of this pathway [108,109]. The mesolimbic system originates in the ventral tegmental area (VTA; A10 dopamine neurons) and projects to limbic targets, most prominently the nucleus accumbens (NAc) [110]. Dopamine (DA) elevations within the NAc are tightly linked to the reinforcing properties of both natural rewards, such as food [111], and drugs of abuse [112], as well as to direct electrical stimulation of the medial forebrain bundle [113]. Significantly, this circuitry not only mediates the primary reinforcing effects of drugs and rewards but also supports associative learning. Environmental and contextual cues paired with drug use acquire secondary reinforcing properties [114,115,116], which can strongly precipitate relapse by eliciting craving and reinstating drug-seeking behavior [112,117]. Transient, phasic dopamine events encode both primary rewards and predictive cues [112,118], highlighting dopamine's dual role in reinforcement and learning. Conversely, the negative affective state during drug withdrawal is associated with a downregulation of mesolimbic dopamine signaling, which further drives compulsive drug-seeking as a form of negative reinforcement [119,120,121].
eCB Influence on Dopamine Transmission
Exogenous cannabinoids, such as Δ9-THC, reliably elevate extracellular DA levels in the ventral striatum [122,123,124]. This dopaminergic activation is mediated by CB1 receptor signaling, as pretreatment with the CB1 antagonist/inverse agonist SR141716A (rimonabant) abolishes Δ9-THC-induced increases in striatal DA [124]. Electrophysiological studies further demonstrate that cannabinoids enhance DA release in the NAc by increasing both the tonic firing rate and burst frequency of midbrain DA neurons [125], an effect reversed by CB1 blockade [126]. Beyond plant-derived cannabinoids, eCBs such as 2-AG can also promote DA neuron excitability, potentially through direct interactions with specific ion channels [127]. However, further in vivo studies are required to establish how this mechanism contributes to reward processing and reinforcement learning. A striking feature of cannabinoid action is that midbrain DA neurons themselves do not express CB1 receptors, implying that cannabinoids regulate DA activity indirectly through local circuit mechanisms. The VTA, the origin of mesolimbic DA projections, is composed of ~60% DA neurons, ~30% GABAergic neurons, and a smaller population of glutamatergic neurons (~3%) [128,129]. In addition to this intrinsic heterogeneity, the VTA receives extensive glutamatergic and GABAergic afferent inputs from limbic and sensory regions, many of which express CB1 receptors [130]. Thus, cannabinoid signaling at these presynaptic sites is well positioned to regulate DA neuron output dynamically.
One of the best-characterized mechanisms involves eCB-mediated modulation of GABAergic control over VTA DA neurons. In vitro application of the GABAA receptor antagonist bicuculline induces robust burst firing of VTA DA neurons, highlighting the powerful inhibitory influence of tonic GABA input on DA excitability [131]. Consistently, administration of the synthetic CB1/CB2 agonist WIN 55,212-2 decreases electrically evoked GABAA-mediated inhibitory postsynaptic currents (IPSCs) in VTA slices. This effect is prevented by CB1 antagonism [132]. These findings suggest that cannabinoids promote DA activity by reducing presynaptic GABAergic inhibition. Moreover, VTA DA neurons themselves can engage in activity-dependent “on-demand” synthesis and release of eCBs, which act retrogradely on CB1 receptors located on local GABA terminals. This feedback loop effectively disinhibits DA neurons, further amplifying phasic activity [130,133]. Cannabinoid modulation of DA signaling is not restricted to the VTA but also extends to its projection targets. In the NAc, CB1 receptors are expressed on both GABAergic and glutamatergic terminals, exerting bidirectional control over DA transmission. Activation of CB1 receptors on GABA terminals enhances DA release within the NAc and facilitates reward-related behaviors [134].
In addition to eCBs acting on the level of dopamine cell bodies within the VTA to interfere with primary and secondary reinforcement processes, they also act on projection sites within the NAc. This interaction involves medium spiny neurons (MSNs) and prefrontal glutamate afferents, particularly focusing on glutamate release at the prelimbic cortex–NAc synapses [146]. Stimulation of these prefrontal glutamate afferents can cause long-term depression (LTD) of NAc glutamatergic synapses, an effect mediated also by 2-AG release and presynaptic CB1-receptor activation [138,147,148]. This form of endocannabinoid-mediated synaptic plasticity in the NAc depends on postsynaptic metabotropic glutamate receptor 5 (mGluR5). In mice, conditional ablation of mGluR5 in dopamine D1-receptor– but not D2-receptor–expressing MSNs (D1 or D2-MSN) by cell-type specific RNA interference [149] abolishes 2-AG-dependent LTD and prevents the expression of drug, natural reward, and brain stimulation–seeking behavior [138]. Pharmacological elevation of 2-AG within the NAc restores both endocannabinoid-dependent-LTD and reward-seeking behavior in these conditional mice [138]. These data expand the disinhibition model, showing that the interaction between endocannabinoids and glutamate within the NAc also contributes to reward-seeking responses [138]. Conversely, activation of CB1 receptors on glutamatergic or cholinergic inputs can decrease DA levels [150]. This dichotomy highlights the circuit- and cell-type-specificity of cannabinoid actions within mesolimbic networks. Taken together, these findings establish that both exogenous and endogenous cannabinoids influence DA transmission predominantly through presynaptic modulation of inhibitory and excitatory inputs onto midbrain DA neurons and their terminals. By orchestrating a fine-tuned balance between disinhibition and excitation across VTA–NAc circuits, cannabinoid signaling provides a powerful mechanism for shaping reward processing, reinforcement, and ultimately, vulnerability to addictive behaviors (Figure 4).
3. Drugs of Abuse and Alcohol-induced eCBS Contribution to Neurodegenerative Disorders
Neurodegenerative (ND) disorders are among the leading causes of disability and mortality worldwide [151,152,153]. These conditions are characterized by the progressive loss of neurons, leading to impaired synaptic connectivity, neuronal death, and cognitive decline. Despite decades of intensive basic and clinical research, current therapies remain largely symptomatic rather than disease-modifying. This limitation likely reflects the slow and often asymptomatic progression of neurodegeneration, which can span decades before clinical symptoms become evident. Recent advances in molecular profiling and neuroimaging have enabled detailed mapping of signaling and transcriptomic alterations in postmortem human brains and in cellular and animal models of ND disorder. These studies have identified both classical pathogenic mechanisms and novel therapeutic targets, including synaptic degeneration—a hallmark shared across multiple ND conditions. Although large-scale genetic studies have revealed several causative mutations, a significant proportion of ND cases lack a clear genetic basis [154]. Increasing evidence implicates environmental factors—such as toxin exposure, nutritional deficiencies, psychosocial stress, and chronic alcohol or drug use—as major contributors to neurodegeneration and its associated behavioral and pathological outcomes. Despite extensive efforts, most pharmacological strategies have demonstrated limited efficacy [155,156], underscoring the urgent need for innovative therapeutic approaches capable of halting or reversing disease progression.
Over the past two decades, considerable progress has been made in elucidating the molecular underpinnings of ND disorders. Nonetheless, these disorders remain devastating and incurable, highlighting the necessity for novel and effective interventions. Emerging evidence positions the eCBS as a pivotal regulator of neuronal function, plasticity, and survival, thereby representing an attractive target for therapeutic development. Dysregulation of eCB signaling has been increasingly linked to the pathophysiology of major ND disorders, including Alzheimer’s disease (AD), Parkinson’s disease (PD), Huntington’s disease (HD), and amyotrophic lateral sclerosis (ALS) [151,157,158]. Moreover, chronic exposure to alcohol and other substances of abuse—such as methamphetamine, opioids, cannabis, and cocaine—induces neuropathological changes that resemble those seen in classical ND disorders [159,160,161,162,163,164]. These substances disrupt eCBS homeostasis, contributing to synaptic dysfunction, neuroinflammation, and neuronal loss that mirror key features of AD, PD, and related conditions. Building on these findings, this section of the review highlights emerging insights into how drug-induced alterations in the eCBS contribute to the onset and progression of cognitive decline and ND diseases, offering new perspectives for therapeutic intervention.
Heavy alcohol consumption remains a major global health concern associated with increased morbidity, mortality, and cognitive impairment. Its long-term impact on dementia-related neuropathology, however, remains unclear. Evidence suggests that alcohol abuse, particularly during adolescence, contributes to persistent behavioral and cognitive deficits. For instance, binge-like alcohol consumption impairs adult cognitive performance in rodent models, as demonstrated by deficits in the novel object recognition, Hebb–Williams, and Morris water maze tasks [165,166,167,168,169]. Chronic alcohol exposure during adolescence appears to be more detrimental than similar exposure during adulthood, producing greater long-term cognitive decline [170,171]. These findings suggest that both the pattern and developmental timing of alcohol use are critical determinants of its deleterious effects on adult and aging-related cognitive function. While some studies report that moderate alcohol consumption is associated with better cognitive performance [172,173,174] or no clear relationship [175,176], heavy drinking is consistently linked to cognitive decline [177,178]. Structural neuroimaging studies have yielded mixed findings: moderate alcohol use has been associated with β-amyloid (Aβ) deposition in middle-aged and older adults without alcohol use disorder (AUD) or dementia [179], whereas other studies found no significant differences in Aβ levels among individuals with AUD [180]. Epidemiological data indicate that older adults (>65 years) who consume more than 14 alcoholic drinks per week have an increased risk of AD and all-cause dementia compared with those who drink less [164,181]. Heavy drinking—defined as two or more drinks per day—has also been associated with earlier disease onset, by up to four years, compared to lighter (less than two/day) drinkers [182]. Future research employing diverse animal models, beyond transgenic systems, is crucial to better understand how alcohol exposure across developmental stages influences aging and AD pathology.
Evidence for a direct relationship between alcohol consumption and PD remains inconsistent. Some studies have reported gender differences in risk, with a higher relative risk in men [183]. In contrast, others suggest that chronic alcohol intake increases PD susceptibility [184]. In alcohol-preferring rats, reduced DA overflow and increased α-synuclein (αSYN) accumulation in the nucleus accumbens have been observed, suggesting that αSYN-mediated alterations in dopaminergic neurotransmission may contribute to alcohol-related motor impairments [185]. However, a definitive association between alcohol use and PD risk has yet to be established, warranting further investigation. Similarly, studies on alcohol and ALS have produced inconsistent results, with population-based analyses suggesting no significant effect of alcohol consumption on ALS risk [186]. Evidence for alcohol influence on the development and progression of HD is inadequate, with only anecdotal reports describing delayed HD diagnosis in alcoholic individuals with a family history of the disease [187].
3.1. Alcohol Abuse–Induced eCBS Alterations and Neurodegeneration
Extensive research in rodent models has demonstrated that various forms of alcohol abuse lead to structural and functional deficits in brain regions critical for cognition [148,149,150,151], along with significant alterations in the eCBS [188,189,190,191]. However, only a limited number of studies have examined the relationship between alcohol-induced eCBS dysregulation and ND-related pathology [188,189,191,192,193,194,195,196]. For example, inhibition of FAAH using URB597 attenuates the effects of adolescent alcohol exposure on memory, neuroinflammation, and brain-derived neurotrophic factor (BDNF) levels in adulthood [194]. Likewise, activation of CB2 receptors prevents alcohol-induced impairment of neurogenesis [193], while CB1 receptor blockade or genetic deletion mitigates adult cognitive and transcriptional deficits induced by early alcohol exposure [197,198,199,200,201,202,203,204]. Recent findings from a transgenic AD mouse model amyloid protein precursor/presenilin (APP/PSEN) show that adolescent binge-like alcohol exposure accelerates cognitive decline at 6 and 12 months, accompanied by increased Aβ42 deposition and reduced expression of the 2-AG–2-AG-metabolizing enzyme MAGL [205]. Alcohol exposure also increased CB2 and DAGLα expression in wild-type, but not AD, mice. These studies collectively suggest that alcohol-induced eCBS dysregulation contributes to age-related cognitive decline and dementia-like pathology. Targeting specific eCBS components may therefore offer a promising strategy to prevent or delay AD progression. For instance, FAAH inhibition not only reduces alcohol-related behaviors [189] but also attenuates AD pathology and disease progression in mouse models [206,207,208]. Furthermore, tau accumulation enhances CB2 expression in neurons, contributing to neurodegeneration in a tauopathy model (hTAUP301L) [209]. Given the overlapping hippocampal pathology observed in aging, eCBS alterations, and alcohol use disorders, it is critical to elucidate how alcohol-induced neurobiological changes at distinct developmental stages predispose the brain to pathological aging and dementia.
3.2. Drugs of Abuse–Induced eCBS Alterations and Neurodegeneration
Drugs of abuse exert profound neurotoxic effects that compromise brain health and contribute to various ND disorders. Chronic cocaine exposure, for instance, promotes neuroinflammatory processes through oxidative stress and mitochondrial and endoplasmic reticulum (ER) dysfunction, accompanied by aberrant activation of microglia and astrogliosis. These events collectively trigger excitotoxicity and neuronal injury [210,211]. Cocaine-induced ER stress, autophagy, and inflammatory signaling lead to protein misfolding and aggregation reminiscent of those observed in ND pathologies [212,213,214]. Moreover, cocaine stimulates astrocyte proliferation and neuroinflammation, further exacerbating neurodegenerative cascades [215].
Cocaine exposure also induces hyperphosphorylation of tau protein in the CNS—an event central to AD pathogenesis [216]. The mechanisms underlying cocaine neurotoxicity are multifactorial and involve excessive cytosolic and synaptic DA accumulation, mitochondrial impairment, oxidative stress, neuroinflammation, pro-apoptotic signaling, altered neuronal plasticity, and disrupted neurogenesis. Notably, cocaine damages dopaminergic neurons and increases the risk of motor dysfunction, showing a threefold higher incidence of dopamine-related motor symptoms among users [217,218,219]. α-Syn directly interacts with and functionally couples to the dopamine transporter (DAT), enhancing DA uptake and accelerating DA-induced apoptosis [220]. Elevated α-syn levels have been observed in the brains of cocaine users, suggesting that increased α-syn production may contribute to PD progression [221]. Consistent with this, Parkinsonian-like motor anomalies have been detected in individuals with a history of cocaine abuse, even after prolonged abstinence [222]. Cocaine abuse has also been associated with aggravated ALS symptoms, suggesting a potential role in disease initiation or acceleration [223]. Although both cocaine [224] and ALS [225] are linked to glutamate-mediated excitotoxicity, the mechanistic connection remains poorly defined and warrants further investigation. Amphetamine-type stimulants (e.g., methamphetamine, MA) likewise induce oxidative stress, apoptosis, and dopaminergic neurodegeneration, leading to behavioral and cognitive impairments [226]. Substantial evidence indicates that MA toxicity drives degeneration of dopaminergic neurons within the nigrostriatal pathway, a hallmark shared with ND disorders such as AD and PD [227,228,229,230]. MA exposure increases the expression of AD-related proteins, including phosphorylated tau and APP [231], and chronic MA users exhibit a heightened risk of PD [232]. Mechanistically, MA induces PD-like pathology via epigenetic upregulation of α-syn expression [233] and by promoting nigrostriatal toxicity through depletion of superoxide dismutase 1 (SOD1) and heightened oxidative stress [194]. Notably, oxidative damage due to SOD1 mutations is also implicated in ALS pathogenesis [194]. Collectively, these observations indicate that, although current studies remain limited, drugs of abuse pose a significant risk for the development and progression of various ND disorders. This highlights the urgent need for systematic investigations to elucidate the mechanistic links between substance abuse and neurodegeneration—two major contributors to morbidity and mortality in the aging population.
Despite the substantial burden imposed by drugs of abuse on the CNS, current pharmacotherapies remain largely ineffective, partly due to an incomplete understanding of the underlying neurobiological mechanisms. The eCBS has emerged as a critical modulator of the neurophysiological and behavioral effects of addictive substances [195,196]. Conversely, drugs of abuse profoundly impact eCBS signaling [108], and eCBS dysregulation has been reported during aging [117] and across multiple ND disorders [111].
Although direct evidence linking drug abuse, eCBS alterations, and ND is limited, several findings highlight eCBS as a promising therapeutic target. For instance, inhibition of FAAH significantly reduces cocaine-induced modulation of CB1 receptor activity [197]. It may confer protection against cocaine-related neurotoxicity [198]. CB1 receptor antagonists attenuate cocaine-induced cellular adaptations, excitatory synaptic alterations, and memory reconsolidation in rodents [199,200], processes critical for cognitive and addictive behaviors. Genetic variations in CNR1 and the FAAH rs324420 polymorphism have been associated with MA abuse. MA exposure also alters eCBS enzyme expression, increasing NAPE-PLD while reducing CB1 receptor and FAAH levels [201], and eCBs mediate MA-induced synaptic activity [202]. The eCBS exerts homeostatic control over neuronal integrity and function throughout aging [111,117,203]. Furthermore, cannabinoids display broad neuroprotective properties against oxidative stress, excitotoxicity, glial reactivity, and protein aggregation—all hallmarks of ND pathology. Moreover, eCBS activity supports adult neurogenesis, a process disrupted in many NDs with prolonged prodromal phases. The localization of eCBS components within key CNS structures that govern neuronal survival and plasticity underscores its potential as an intrinsic defense system against neurotoxic insults, including oxidative stress, protein dysregulation, and inflammation. Experimental evidence supports the neuroprotective capacity of both endogenous cannabinoids and phytocannabinoids (for review, see [203,204,205]). CB2 receptor agonists, in particular, exhibit protective effects against drug-induced neurotoxicity [204] and demonstrate neuroprotection in animal models of AD [205], PD [118,206], HD [207,208], ALS [209,210], and other NDs [111,211]. Although the optimal eCBS targets likely vary by disease type and progression stage, continued investigation into the interplay between drug abuse, eCBS modulation, and neurodegeneration will be essential for the development of targeted neuroprotective therapies.
4. Conclusions
The eCBS occupies a pivotal intersection between synaptic plasticity, addiction, and neurodegeneration. Drugs of abuse and alcohol profoundly disrupt eCB signaling, triggering oxidative, inflammatory, and proteostatic imbalances that accelerate neuronal dysfunction and death. Conversely, the eCBS exerts endogenous neuroprotective actions by regulating neurotransmission, neurogenesis, and glial activity. Evidence from preclinical models highlights that modulating specific eCBS targets—such as CB1, CB2, FAAH, and MAGL—can attenuate drug-induced neurotoxicity and ameliorate neuropathological features of Alzheimer’s, Parkinson’s, Huntington’s, and motor neuron diseases (Figure 5). However, translational studies remain limited. Future research integrating molecular, imaging, and behavioral approaches is essential to delineate the causal pathways linking drug abuse, eCBS dysregulation, and neurodegeneration. Such efforts will advance mechanistic understanding and foster the development of cannabinoid-based or eCBS-targeted interventions aimed at restoring neuronal resilience and preventing neurodegenerative decline across the lifespan.
Author Contributions
Both authors participated in the collection of literature, writing, and editing of the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
National Institute of Alcohol and Alcoholism grant (R01 AA029686) to BSB.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
National Institute of Alcohol and Alcoholism grant (R01 AA029686) to BSB.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
The following abbreviations are used in this manuscript:
| eCBS | endocannabinoid system |
| ND | neurodegenerative |
| CB1 | cannabinoid receptors 1 |
| LD | Linear dichroism |
| CB2 | cannabinoid receptors 2 |
| CNS | central nervous system |
| Δ9-THC | Δ9- tetrahydrocannabinol |
| GPCR | G protein–coupled receptor |
| PFC | prefrontal cortex |
| NAc | nucleus accumbens |
| cAMP | cyclic adenosine monophosphate |
| MAPKs | mitogen-activated protein kinases |
| ERK | extracellular signal-regulated kinases |
| JNK | Jun N-terminal kinase |
| CREB | cAMP-response element binding protein |
| PI3K | phosphatidylinositol 3-kinase |
| AKT | protein kinase B |
| Src | family of non-receptor tyrosine kinases |
| VTA | ventral tegmental area |
| VGCC | voltage-gated calcium channels |
| GIRKs | G protein–coupled inwardly rectifying potassium channels |
| AEA | N-arachidonoylethanolamine |
| 2-AG | 2-arachidonoylglycerol |
| NAPE | N-acyl-phosphatidylethanolamine |
| PLD | phospholipase D |
| DAG | diacylglycerol |
| DAGLα/β | diacylglycerol alpha/ beta |
| FAAH | fatty acid amide hydrolase |
| MAGL | monoacylglycerol lipase |
| COX-2 | cyclooxygenase-2 |
| DA | Dopamine |
| SR141716A | rimonabant |
| GABA | Gamma-Aminobutyric Acid |
| IPSCs | inhibitory postsynaptic currents |
| MSNs | medium spiny neurons |
| mGluR5 | metabotropic glutamate receptor 5 |
| LTD | long-term depression |
| AD | Alzheimer’s disease |
| PD | Parkinson’s disease |
| HD | Huntington’s disease |
| Aβ | β-amyloid |
| AUD | alcohol use disorder |
| ALS | amyotrophic lateral sclerosis |
| BDNF | brain-derived neurotrophic factor |
| APP/ PSEN | Amyloid protein precursor/presenilin |
| DAT | dopamine transporter |
| MA | methamphetamine |
| SOD1 | superoxide dismutase 1 |
| CNR1 | cannabinoid receptor 1 protein coding gene |
| PPARα | proliferator-activated receptor alpha |
| PPARγ | peroxisome proliferator-activated receptor gamma |
References
- Di Marzo, V. , et al., Endocannabinoids:endogenous cannabinoid receptor ligands with neuromodulatory action. Trends Neurosci 1998, 21, 521–528. [Google Scholar] [CrossRef] [PubMed]
- Mechoulam, R. Fride, and V. Di Marzo, Endocannabinoids. Eur J Pharmacol 1998, 359, 1–18. [Google Scholar] [CrossRef]
- Devane, W.A. , et al., Determination and characterization of a cannabinoid receptor in rat brain. Mol. Pharmacol., 1988, 34, 605–613. [Google Scholar] [CrossRef]
- Howlett, A.C. , et al., The cannabinoid receptor: biochemical, anatomical and behavioral characterization. Trends Neurosci 1990, 13, 420–423. [Google Scholar] [CrossRef]
- Devane, W.A. , et al., Isolation and structure of a brain constituent that binds to the cannabinoid receptor. Science., 1992, 258, 1946–1949. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, L.A. , et al., Structure of a cannabinoid receptor and functional expression of the cloned cDNA. Nature, 1990, 346, 561–564. [Google Scholar] [CrossRef]
- Matsuda, L.A., T. I. Bonner, and S.J. Lolait, Localization of cannabinoid receptor mRNA in rat brain. J Comp Neurol, 1993, 327, 535–550. [Google Scholar] [CrossRef] [PubMed]
- Compton, D.R. , et al., Cannabinoid structure-activity relationships: correlation of receptor binding and in vivo activities. J Pharmacol Exp Ther 1993, 265, 218–226. [Google Scholar] [CrossRef]
- Glass, M. L. Faull, and M.Dragunow, Loss of cannabinoid receptors in the substantia nigra in Huntington's disease. Neuroscience 1993, 56, 523–527. [Google Scholar] [CrossRef]
- Melvin, L.S. , et al., Structure-activity relationships for cannabinoid receptor-binding and analgesic activity: studies of bicyclic cannabinoid analogs. Mol Pharmacol 1993, 44, 1008–1015. [Google Scholar] [CrossRef]
- Collins, D.R. G.Pertwee, and S.N. Davies, The action of synthetic cannabinoids on the induction of long-term potentiation in the rat hippocampal slice. Eur J Pharmacol 1994, 259, R7–R8. [Google Scholar] [CrossRef]
- Navarro, M. , et al., Motor behavior and nigrostriatal dopaminergic activity in adult rats perinatally exposed to cannabinoids. Pharmacol Biochem Behav, 1994, 47, 47–58. [Google Scholar] [CrossRef] [PubMed]
- Lichtman, A.H. R.Dimen, and B.R. Martin, Systemic or intrahippocampal cannabinoid administration impairs spatial memory in rats. Psychopharmacology (Berl), 1995, 119, 282–290. [Google Scholar] [CrossRef]
- Munro, S., K. L. Thomas, and M. Abu-Shaar, Molecular characterization of a peripheral receptor for cannabinoids. Nature, 1993, 365, 61–65. [Google Scholar] [CrossRef] [PubMed]
- Breivogel, C.S. and S.R. Childers, Cannabinoid agonist signal transduction in rat brain: comparison of cannabinoid agonists in receptor binding, G-protein activation, and adenylyl cyclase inhibition. J Pharmacol Exp Ther, 2000, 295, 328–336. [Google Scholar] [CrossRef]
- Derkinderen, P. , et al., Regulation of extracellular signal-regulated kinase by cannabinoids in hippocampus. J Neurosci, 2003, 23, 2371–2382. [Google Scholar]
- Mackie, K. , Mechanisms of CB1 receptor signaling: endocannabinoid modulation of synaptic strength. Int J Obes (Lond) 2006, 30 (Suppl 1), S19–23. [Google Scholar] [CrossRef] [PubMed]
- Mechoulam, R. and L.A. Parker, The endocannabinoid system and the brain. Annu Rev Psychol, 2013, 64, 21–47. [Google Scholar] [CrossRef]
- Howlett, A.C. , Pharmacology of cannabinoid receptors. Annu Rev Pharmacol Toxicol, 1995, 35, 607–634. [Google Scholar] [CrossRef]
- Childers, S.R., T. Sexton, and M.B. Roy, Effects of anandamide on cannabinoid receptors in rat brain membranes. Biochem. Pharmacol., 1994, 47, 711–715. [Google Scholar] [CrossRef]
- Pinto, J.C. , et al., Cannabinoid receptor binding and agonist activity of amides and esters of arachidonic acid. Mol. Pharmacol., 1994, 46, 516–522. [Google Scholar] [CrossRef]
- Howlett, A.C. and S. Mukhopadhyay, Cellular signal transduction by anandamide and 2-arachidonoylglycerol. Chem Phys Lipids., 2000, 108, 53–70. [Google Scholar] [CrossRef]
- Caulfield, M.P. and D.A. Brown, Cannabinoid receptor agonists inhibit Ca current in NG108-15 neuroblastoma cells via a pertussis toxin-sensitive mechanism. Br J Pharmacol, 1992, 106, 231–232. [Google Scholar] [CrossRef]
- Mackie, K. and B. Hille, Cannabinoids inhibit N-type calcium channels in neuroblastoma-glioma cells. Proc Natl Acad Sci U S A, 1992, 89, 3825–3829. [Google Scholar] [CrossRef]
- Nogueron, M.I. , et al., Cannabinoid receptor agonists inhibit depolarization-induced calcium influx in cerebellar granule neurons. J Neurochem, 2001, 79, 371–381. [Google Scholar] [CrossRef] [PubMed]
- Pan, X., S. R. Ikeda, and D.L. Lewis, Rat brain cannabinoid receptor modulates N-type Ca2+ channels in a neuronal expression system. Mol Pharmacol., 1996, 49, 707–714. [Google Scholar] [CrossRef] [PubMed]
- Mu, J. , et al., Cannabinoid receptors differentially modulate potassium A and D currents in hippocampal neurons in culture. J. Pharmacol. Exp. Ther., 1999, 291, 893–902. [Google Scholar] [CrossRef] [PubMed]
- Freund, T.F., I. Katona, and D. Piomelli, Role of endogenous cannabinoids in synaptic signaling. Physiol Rev., 2003, 83, 1017–1066. [Google Scholar] [CrossRef]
- Howlett, A.C. , et al., International Union of Pharmacology. XXVII. Classification of cannabinoid receptors. Pharmacol Rev, 2002, 54, 161–202. [Google Scholar] [CrossRef]
- Graham, E.S. , et al., Induction of Krox-24 by endogenous cannabinoid type 1 receptors in Neuro2A cells is mediated by the MEK-ERK MAPK pathway and is suppressed by the phosphatidylinositol 3-kinase pathway. J Biol Chem 2006, 281, 29085–29095. [Google Scholar] [CrossRef]
- Basavarajappa, B.S. and O. Arancio, Synaptic Plasticity: Emerging Role for Endocannabinoid system, in Synaptic Plasticity: New Research, T.F. Kaiser and F.J. Peters, Editors. 2008, Nova Science Publishers, Inc.: NY, USA. p. pp77-112.
- Ozaita, A. Puighermanal, and R.Maldonado, Regulation of PI3K/Akt/GSK-3 pathway by cannabinoids in the brain. J Neurochem 2007, 102, 1105–1114. [Google Scholar] [CrossRef] [PubMed]
- Basavarajappa, B.S. , Fetal Alcohol Spectrum Disorder: Potential Role of Endocannabinoids Signaling. Brain Sci, 2015, 5, 456–493. [Google Scholar] [CrossRef]
- Basavarajappa, B.S. and S.Subbanna, CB1 Receptor-Mediated Signaling Underlies the Hippocampal Synaptic, Learning and Memory Deficits Following Treatment with JWH-081, a New Component of Spice/K2 Preparations. Hippocampus 2014, 24, 178–188. [Google Scholar] [CrossRef]
- Bayewitch, M. , et al., The peripheral cannabinoid receptor: adenylate cyclase inhibition and G protein coupling. FEBS Lett 1995, 375, 143–147. [Google Scholar] [CrossRef]
- Egertova, M. , et al., A new perspective on cannabinoid signalling: complementary localization of fatty acid amide hydrolase and the CB1 receptor in rat brain. Proc R Soc Lond B Biol Sci, 1998, 265, 2081–2085. [Google Scholar] [CrossRef]
- Tsou, K. , et al., Immunohistochemical distribution of cannabinoid CB1 receptors in the rat central nervous system. Neuroscience, 1998, 83, 393–411. [Google Scholar] [CrossRef]
- Marsicano, G. and B.Lutz, Expression of the cannabinoid receptor CB1 in distinct neuronal subpopulations in the adult mouse forebrain. Eur J Neurosci 1999, 11, 4213–4225. [Google Scholar] [CrossRef]
- Terzian, A.L. Micale, and C.T. Wotjak, Cannabinoid receptor type 1 receptors on GABAergic vs. glutamatergic neurons differentially gate sex-dependent social interest in mice. Eur J Neurosci 2014, 40, 2293–2298. [Google Scholar] [CrossRef]
- Haring, M. , et al., Identification of the cannabinoid receptor type 1 in serotonergic cells of raphe nuclei in mice. Neuroscience 2007, 146, 1212–1219. [Google Scholar] [CrossRef] [PubMed]
- Oropeza, V.C. Mackie, and E.J. Van Bockstaele, Cannabinoid receptors are localized to noradrenergic axon terminals in the rat frontal cortex. Brain Res 2007, 1127, 36–44. [Google Scholar] [CrossRef] [PubMed]
- Mackie, K. , Cannabinoid receptor homo- and heterodimerization. Life Sci, 2005, 77, 1667–1673. [Google Scholar] [CrossRef]
- Navarrete, M. Diez, and A.Araque, Astrocytes in endocannabinoid signalling. Philos Trans R Soc Lond B Biol Sci, 2014, 369, 20130599. [Google Scholar] [CrossRef]
- Cota, D. , et al., The endogenous cannabinoid system affects energy balance via central orexigenic drive and peripheral lipogenesis. J Clin Invest, 2003, 112, 423–431. [Google Scholar]
- Parolaro, D. , Presence and functional regulation of cannabinoid receptors in immune cells. Life Sci, 1999, 65, 637–644. [Google Scholar] [CrossRef] [PubMed]
- Bouaboula, M. , et al., Activation of mitogen-activated protein kinases by stimulation of the central cannabinoid receptor CB1. Biochem J 1995, 312 Pt 2, 637–641. [Google Scholar] [CrossRef] [PubMed]
- Cuddihey, H. K.MacNaughton, and K.A. Sharkey, Role of the Endocannabinoid System in the Regulation of Intestinal Homeostasis. Cell Mol Gastroenterol Hepatol, 2022, 14, 947–963. [Google Scholar] [CrossRef]
- Howlett, A.C. and M.E. Abood, CB(1) and CB(2) Receptor Pharmacology. Adv Pharmacol, 2017, 80, 169–206. [Google Scholar]
- Rezq, S. Hassan, and M.F. Mahmoud, Rimonabant ameliorates hepatic ischemia/reperfusion injury in rats: Involvement of autophagy via modulating ERK- and PI3K/AKT-mTOR pathways. Int Immunopharmacol, 2021, 100, 108140. [Google Scholar] [CrossRef] [PubMed]
- Rorabaugh, B.R. and D.J. Morgan, CB1 receptor coupling to extracellular regulated kinase via multiple Galphai/o isoforms. Neuroreport, 2025, 36, 191–195. [Google Scholar] [CrossRef]
- Scherma, M. , et al., Brain activity of anandamide: a rewarding bliss? Acta Pharmacol Sin, 2019, 40, 309–323. [Google Scholar] [CrossRef]
- Song, L. , et al., The CB1 cannabinoid receptor agonist reduces L-DOPA-induced motor fluctuation and ERK1/2 phosphorylation in 6-OHDA-lesioned rats. Drug Des Devel Ther, 2014, 8, 2173–2179. [Google Scholar] [PubMed]
- Subbanna, S. , et al., Ethanol exposure induces neonatal neurodegeneration by enhancing CB1R Exon1 histone H4K8 acetylation and up-regulating CB1R function causing neurobehavioral abnormalities in adult mice. Int J Neuropsychopharmacol, 2015, 18, 1–15. [Google Scholar] [CrossRef]
- Subbanna, S. , et al., Anandamide-CB1 Receptor Signaling Contributes to Postnatal Ethanol-Induced Neonatal Neurodegeneration, Adult Synaptic and Memory Deficits. Journal of neuoscience, 2013, 33, 6350–6366. [Google Scholar]
- Rueda, D. , et al., The endocannabinoid anandamide inhibits neuronal progenitor cell differentiation through attenuation of the Rap1/B-Raf/ERK pathway. J Biol Chem, 2002, 277, 46645–46650. [Google Scholar] [CrossRef]
- Gomez del Pulgar, T., G. Velasco, and M. Guzman, The CB1 cannabinoid receptor is coupled to the activation of protein kinase B/Akt. Biochem J., 2000, 347, 369–373. [Google Scholar] [CrossRef]
- Di Scala, C. , et al., Anandamide Revisited: How Cholesterol and Ceramides Control Receptor-Dependent and Receptor-Independent Signal Transmission Pathways of a Lipid Neurotransmitter. Biomolecules, 2018, 8.
- Diana, M.A. and A.Marty, Endocannabinoid-mediated short-term synaptic plasticity: depolarization-induced suppression of inhibition (DSI) and depolarization-induced suppression of excitation (DSE). Br J Pharmacol, 2004, 142, 9–19. [Google Scholar] [CrossRef]
- De Waard, M. , et al., Direct binding of G-protein betagamma complex to voltage-dependent calcium channels. Nature 1997, 385, 446–450. [Google Scholar] [CrossRef]
- De Waard, M. , et al., How do G proteins directly control neuronal Ca2+ channel function? Trends Pharmacol Sci, 2005, 26, 427–436. [Google Scholar] [CrossRef]
- Khan, S.M. Y.Sung, and T.E. Hebert, Gbetagamma subunits-Different spaces, different faces. Pharmacol Res, 2016, 111, 434–441. [Google Scholar] [CrossRef] [PubMed]
- Bouaboula, M. , et al., Signaling pathway associated with stimulation of CB2 peripheral cannabinoid receptor. Involvement of both mitogen-activated protein kinase and induction of Krox-24 expression. Eur J Biochem, 1996, 237, 704–711. [Google Scholar] [CrossRef] [PubMed]
- Maresz, K. , et al., Modulation of the cannabinoid CB2 receptor in microglial cells in response to inflammatory stimuli. J Neurochem, 2005, 95, 437–445. [Google Scholar] [CrossRef]
- Gong, J.P. , et al., Cannabinoid CB2 receptors: immunohistochemical localization in rat brain. Brain Res, 2006, 1071, 10–23. [Google Scholar] [CrossRef]
- Onaivi, E.S. , et al., Discovery of the presence and functional expression of cannabinoid CB2 receptors in brain. Ann N Y Acad Sci, 2006, 1074, 514–536. [Google Scholar] [CrossRef] [PubMed]
- Xin, Q. , et al., The impact of cannabinoid type 2 receptors (CB2Rs) in neuroprotection against neurological disorders. Acta Pharmacol Sin, 2020, 41, 1507–1518. [Google Scholar] [CrossRef]
- Yu, X. Jia, and Y.Dong, Research progress on the cannabinoid type-2 receptor and Parkinson's disease. Front Aging Neurosci, 2023, 15, 1298166. [Google Scholar] [CrossRef]
- Li, Y. and J.Kim, Neuronal expression of CB2 cannabinoid receptor mRNAs in the mouse hippocampus. Neuroscience 2015, 311, 253–267. [Google Scholar] [CrossRef]
- Stempel, A.V. , et al., Cannabinoid Type 2 Receptors Mediate a Cell Type-Specific Plasticity in the Hippocampus. Neuron, 2016, 90, 795–809. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.Y. , et al., Expression of functional cannabinoid CB(2) receptor in VTA dopamine neurons in rats. Addict Biol, 2017, 22, 752–765. [Google Scholar] [CrossRef]
- Slipetz, D.M. , et al., Activation of the human peripheral cannabinoid receptor results in inhibition of adenylyl cyclase. Mol Pharmacol, 1995, 48, 352–361. [Google Scholar] [CrossRef]
- Shoemaker, J.L. , et al., Agonist-directed trafficking of response by endocannabinoids acting at CB2 receptors. J Pharmacol Exp Ther, 2005, 315, 828–838. [Google Scholar] [CrossRef] [PubMed]
- Felder, C.C. , et al., Comparison of the pharmacology and signal transduction of the human cannabinoid CB1 and CB2 receptors. Mol Pharmacol, 1995, 48, 443–450. [Google Scholar] [CrossRef]
- Kobayashi, Y. , et al., Activation by 2-arachidonoylglycerol, an endogenous cannabinoid receptor ligand, of p42/44 mitogen-activated protein kinase in HL-60 cells. J Biochem 2001, 129, 665–669. [Google Scholar] [CrossRef]
- Molina-Holgado, E. , et al., Cannabinoids promote oligodendrocyte progenitor survival: involvement of cannabinoid receptors and phosphatidylinositol-3 kinase/Akt signaling. J Neurosci, 2002, 22, 9742–9753. [Google Scholar] [CrossRef] [PubMed]
- Patel, M. , et al., Delineating the interactions between the cannabinoid CB(2) receptor and its regulatory effectors; beta-arrestins and GPCR kinases. Br J Pharmacol, 2022, 179, 2223–2239. [Google Scholar] [CrossRef] [PubMed]
- Morales-Pastor, A. , et al., Multiple intramolecular triggers converge to preferential G protein coupling in the CB(2)R. Nat Commun, 2025, 16, 5265. [Google Scholar] [CrossRef]
- McAllister, S.D. , et al., Cannabinoid receptors can activate and inhibit G protein-coupled inwardly rectifying potassium channels in a xenopus oocyte expression system. J Pharmacol Exp Ther, 1999, 291, 618–626. [Google Scholar] [CrossRef]
- Ho, B.Y. , et al., Coupling of the expressed cannabinoid CB1 and CB2 receptors to phospholipase C and G protein-coupled inwardly rectifying K+ channels. Recept Channels, 1999, 6, 363–374. [Google Scholar]
- Soobben, M. Sayed, and I.Achilonu, Exploring the evolutionary trajectory and functional landscape of cannabinoid receptors: A comprehensive bioinformatic analysis. Comput Biol Chem, 2024, 112, 108138. [Google Scholar] [CrossRef]
- Bala, K. Porel, and K.R. Aran, Emerging roles of cannabinoid receptor CB2 receptor in the central nervous system: therapeutic target for CNS disorders. Psychopharmacology (Berl), 2024, 241, 1939–1954. [Google Scholar] [CrossRef] [PubMed]
- Pertwee, R.G. , Pharmacology of cannabinoid CB1 and CB2 receptors. Pharmacol Ther., 1997, 74, 129–180. [Google Scholar] [CrossRef]
- Buckley, N.E. , et al., Expression of the CB1 and CB2 receptor messenger RNAs during embryonic development in the rat. Neuroscience, 1998, 82, 1131–1149. [Google Scholar] [CrossRef]
- Carracedo, A. , et al., Cannabinoids induce apoptosis of pancreatic tumor cells via endoplasmic reticulum stress-related genes. Cancer Res, 2006, 66, 6748–6755. [Google Scholar] [CrossRef]
- Carracedo, A. , et al., The stress-regulated protein p8 mediates cannabinoid-induced apoptosis of tumor cells. Cancer Cell 2006, 9, 301–312. [Google Scholar] [CrossRef]
- Carrier, E.J. , et al., Cultured rat microglial cells synthesize the endocannabinoid 2-arachidonylglycerol, which increases proliferation via a CB2 receptor-dependent mechanism. Mol Pharmacol, 2004, 65, 999–1007. [Google Scholar] [CrossRef]
- Choi, I.Y. , et al., Activation of cannabinoid CB2 receptor-mediated AMPK/CREB pathway reduces cerebral ischemic injury. Am J Pathol, 2013, 182, 928–939. [Google Scholar] [CrossRef]
- Gertsch, J. , et al., Echinacea alkylamides modulate TNF-alpha gene expression via cannabinoid receptor CB2 and multiple signal transduction pathways. FEBS Lett, 2004, 577, 563–569. [Google Scholar] [CrossRef]
- Herrera, B. , et al., p38 MAPK is involved in CB2 receptor-induced apoptosis of human leukaemia cells. FEBS Lett, 2005, 579, 5084–5088. [Google Scholar] [CrossRef]
- Palazuelos, J. , et al., Non-psychoactive CB2 cannabinoid agonists stimulate neural progenitor proliferation. FASEB J, 2006, 20, 2405–2407. [Google Scholar] [CrossRef] [PubMed]
- Samson, M.T. , et al., Differential roles of CB1 and CB2 cannabinoid receptors in mast cells. J Immunol, 2003, 170, 4953–4962. [Google Scholar] [CrossRef] [PubMed]
- Mechoulam, R. , et al., Identification of an endogenous 2-monoglyceride, present in canine gut, that binds to cannabinoid receptors. Biochem Pharmacol 1995, 50, 83–90. [Google Scholar] [CrossRef] [PubMed]
- Sugiura, T. , et al., 2-Arachidonoylglycerol: a possible endogenous cannabinoid receptor ligand in brain. Biochem Biophys Res Commun, 1995, 215, 89–97. [Google Scholar] [CrossRef]
- Di Marzo, V. and L. De Petrocellis, Why do cannabinoid receptors have more than one endogenous ligand? Philos Trans R Soc Lond B Biol Sci, 2012, 367, 3216–3228. [Google Scholar] [CrossRef]
- Pertwee, R.G. , et al., International Union of Basic and Clinical Pharmacology. LXXIX. Cannabinoid receptors and their ligands: beyond CB(1) and CB(2). Pharmacol Rev, 2010, 62, 588–631. [Google Scholar] [CrossRef]
- Lisboa, S.F. and F.S. Guimaraes, Differential role of CB1 and TRPV1 receptors on anandamide modulation of defensive responses induced by nitric oxide in the dorsolateral periaqueductal gray. Neuropharmacology 2012, 62, 2455–2462. [Google Scholar] [CrossRef]
- O'Sullivan, S.E. , An update on PPAR activation by cannabinoids. Br J Pharmacol, 2016, 173, 1899–1910. [Google Scholar] [CrossRef]
- Kano, M. , et al., Endocannabinoid-mediated control of synaptic transmission. Physiol Rev, 2009, 89, 309–380. [Google Scholar] [CrossRef] [PubMed]
- Katona, I. , et al., Presynaptically located CB1 cannabinoid receptors regulate GABA release from axon terminals of specific hippocampal interneurons. J Neurosci, 1999, 19, 4544–4558. [Google Scholar] [CrossRef] [PubMed]
- Alger, B.E. , Retrograde signaling in the regulation of synaptic transmission: focus on endocannabinoids. Progress in Neurobiology., 2002, 68, 247–286. [Google Scholar] [CrossRef]
- Lutz, B. , et al., The endocannabinoid system in guarding against fear, anxiety and stress. Nat Rev Neurosci, 2015, 16, 705–718. [Google Scholar] [PubMed]
- Liu, J. , et al., A biosynthetic pathway for anandamide. Proc Natl Acad Sci U S A, 2006, 103, 13345–13350. [Google Scholar] [CrossRef]
- Murataeva, N. Straiker, and K.Mackie, Parsing the players: 2-arachidonoylglycerol synthesis and degradation in the CNS. Br J Pharmacol 2014, 171, 1379–1391. [Google Scholar] [CrossRef]
- Basavarajappa, B.S. , Critical Enzymes Involved in Endocannabinoid Metabolism. Protein and Peptide letters, 2007, 14, 237–246. [Google Scholar] [CrossRef] [PubMed]
- Basavarajappa, B.S. , Major Enzymes of Endocannabinoid Metabolism, in Frontiers in Protein and Peptide Sciences, B. Dunn, Editor. 2014, Bentham Science Publishers: Oak Park, IL, USA. p. 31-62.
- Rouzer, C.A. and L.J.Marnett, Endocannabinoid oxygenation by cyclooxygenases, lipoxygenases, and cytochromes P450: cross-talk between the eicosanoid and endocannabinoid signaling pathways. Chem Rev, 2011, 111, 5899–5921. [Google Scholar] [CrossRef]
- Ligresti, A., L. De Petrocellis, and V. Di Marzo, From Phytocannabinoids to Cannabinoid Receptors and Endocannabinoids: Pleiotropic Physiological and Pathological Roles Through Complex Pharmacology. Physiol Rev, 2016, 96, 1593–1659. [Google Scholar] [PubMed]
- Di Chiara, G. and A.Imperato, Drugs abused by humans preferentially increase synaptic dopamine concentrations in the mesolimbic system of freely moving rats. Proc Natl Acad Sci U S A, 1988, 85, 5274–5278. [Google Scholar] [CrossRef] [PubMed]
- Pierce, R.C. and V. Kumaresan, The mesolimbic dopamine system: the final common pathway for the reinforcing effect of drugs of abuse? Neurosci Biobehav Rev, 2006, 30, 215–238. [Google Scholar] [CrossRef]
- Hillarp, N.A. Fuxe, and A.Dahlstrom, Demonstration and mapping of central neurons containing dopamine, noradrenaline, and 5-hydroxytryptamine and their reactions to psychopharmaca. Pharmacol Rev, 1966, 18, 727–741. [Google Scholar] [CrossRef]
- Roitman, M.F. , et al., Dopamine operates as a subsecond modulator of food seeking. J Neurosci, 2004, 24, 1265–1271. [Google Scholar]
- Phillips, P.E. , et al., Subsecond dopamine release promotes cocaine seeking. Nature, 2003, 422, 614–618. [Google Scholar]
- Cheer, J.F. , et al., Phasic dopamine release evoked by abused substances requires cannabinoid receptor activation. J Neurosci, 2007, 27, 791–795. [Google Scholar]
- Berridge, K.C. and T.E. Robinson, What is the role of dopamine in reward: hedonic impact, reward learning, or incentive salience? Brain Res Brain Res Rev 1998, 28, 309–369. [Google Scholar] [CrossRef] [PubMed]
- Bindra, D. , Neuropsychological interpretation of the effects of drive and incentive-motivation on general and instrumental behavior. Psychol Rev 1968, 75, 1–22. [Google Scholar] [CrossRef]
- Flagel, S.B. , et al., A selective role for dopamine in stimulus-reward learning. Nature 2011, 469, 53–57. [Google Scholar]
- Nicola, S.M. , The flexible approach hypothesis: unification of effort and cue-responding hypotheses for the role of nucleus accumbens dopamine in the activation of reward-seeking behavior. J Neurosci, 2010, 30, 16585–16600. [Google Scholar] [CrossRef]
- Owesson-White, C.A. , et al., Neural encoding of cocaine-seeking behavior is coincident with phasic dopamine release in the accumbens core and shell. Eur J Neurosci, 2009, 30, 1117–1127. [Google Scholar]
- Koob, G.F. , Neurobiological substrates for the dark side of compulsivity in addiction. Neuropharmacology, 2009, 56 Suppl 1, 18-31.
- Weiss, F. , et al., Compulsive drug-seeking behavior and relapse. Neuroadaptation, stress, and conditioning factors. Ann N Y Acad Sci, 2001, 937, 1–26. [Google Scholar]
- Wenzel, J.M. , et al., Phasic Dopamine Signals in the Nucleus Accumbens that Cause Active Avoidance Require Endocannabinoid Mobilization in the Midbrain. Curr Biol, 2018, 28, 1392–1404. [Google Scholar] [PubMed]
- Cheer, J.F. , et al., Cannabinoids enhance subsecond dopamine release in the nucleus accumbens of awake rats. J Neurosci, 2004, 24, 4393–4400. [Google Scholar]
- Chen, J.P. , et al., Delta 9-tetrahydrocannabinol produces naloxone-blockable enhancement of presynaptic basal dopamine efflux in nucleus accumbens of conscious, freely-moving rats as measured by intracerebral microdialysis. Psychopharmacology (Berl), 1990, 102, 156–162. [Google Scholar]
- Tanda, G. E.Pontieri, and G. Di Chiara, Cannabinoid and heroin activation of mesolimbic dopamine transmission by a common mu1 opioid receptor mechanism. Science, 1997, 276, 2048–2050. [Google Scholar] [CrossRef]
- French, E.D. , delta9-Tetrahydrocannabinol excites rat VTA dopamine neurons through activation of cannabinoid CB1 but not opioid receptors. Neurosci Lett, 1997, 226, 159–162. [Google Scholar] [CrossRef]
- Gessa, G.L. , et al., Cannabinoids activate mesolimbic dopamine neurons by an action on cannabinoid CB1 receptors. Eur J Pharmacol, 1998, 341, 39–44. [Google Scholar]
- Gantz, S.C. and B.P. Bean, Cell-Autonomous Excitation of Midbrain Dopamine Neurons by Endocannabinoid-Dependent Lipid Signaling. Neuron 2017, 93, 1375–1387. [Google Scholar] [CrossRef]
- Dobi, A. , et al., Glutamatergic and nonglutamatergic neurons of the ventral tegmental area establish local synaptic contacts with dopaminergic and nondopaminergic neurons. J Neurosci, 2010, 30, 218–229. [Google Scholar] [PubMed]
- Swanson, L.W. , The projections of the ventral tegmental area and adjacent regions: a combined fluorescent retrograde tracer and immunofluorescence study in the rat. Brain Res Bull, 1982, 9, 321–353. [Google Scholar] [CrossRef] [PubMed]
- Riegel, A.C. and C.R. Lupica, Independent presynaptic and postsynaptic mechanisms regulate endocannabinoid signaling at multiple synapses in the ventral tegmental area. J Neurosci, 2004, 24, 11070–11078. [Google Scholar] [CrossRef]
- Cheer, J.F. , et al., Lack of response suppression follows repeated ventral tegmental cannabinoid administration: an in vitro electrophysiological study. Neuroscience, 2000, 99, 661–667. [Google Scholar]
- Szabo, B. Siemes, and I.Wallmichrath, Inhibition of GABAergic neurotransmission in the ventral tegmental area by cannabinoids. Eur J Neurosci, 2002, 15, 2057–2061. [Google Scholar] [CrossRef] [PubMed]
- Lupica, C.R. and A.C. Riegel, Endocannabinoid release from midbrain dopamine neurons: a potential substrate for cannabinoid receptor antagonist treatment of addiction. Neuropharmacology 2005, 48, 1105–1116. [Google Scholar] [CrossRef]
- Sperlagh, B. , et al., Neurochemical evidence that stimulation of CB1 cannabinoid receptors on GABAergic nerve terminals activates the dopaminergic reward system by increasing dopamine release in the rat nucleus accumbens. Neurochem Int, 2009, 54, 452–457. [Google Scholar]
- Li, X. , et al., Crystal Structure of the Human Cannabinoid Receptor CB2. Cell, 2019, 176, 459–467. [Google Scholar]
- Augier, E. , et al., The GABA(B) Positive Allosteric Modulator ADX71441 Attenuates Alcohol Self-Administration and Relapse to Alcohol Seeking in Rats. Neuropsychopharmacology, 2017, 42, 1789–1799. [Google Scholar]
- van den Brink, W. , et al., Efficacy and safety of sodium oxybate in alcohol-dependent patients with a very high drinking risk level. Addict Biol, 2018, 23, 969–986. [Google Scholar]
- Bilbao, A. , et al., Endocannabinoid LTD in Accumbal D1 Neurons Mediates Reward-Seeking Behavior. iScience, 2020, 23, 100951. [Google Scholar]
- Wang, X. , et al., Role of mGluR5 neurotransmission in reinstated cocaine-seeking. Addict Biol, 2013, 18, 40–49. [Google Scholar] [PubMed]
- Olive, M.F. Metabotropic glutamate receptor ligands as potential therapeutics for addiction. Curr Drug Abuse Rev, 2009, 2, 83–98. [Google Scholar] [CrossRef] [PubMed]
- Chen, M. , et al., Morphine disinhibits glutamatergic input to VTA dopamine neurons and promotes dopamine neuron excitation. Elife, 2015, 4. [Google Scholar]
- Laaris, N. H.Good, and C.R. Lupica, Delta9-tetrahydrocannabinol is a full agonist at CB1 receptors on GABA neuron axon terminals in the hippocampus. Neuropharmacology, 2010, 59, 121–127. [Google Scholar] [CrossRef]
- Shen, M. and S.A. Thayer, Delta9-tetrahydrocannabinol acts as a partial agonist to modulate glutamatergic synaptic transmission between rat hippocampal neurons in culture. Mol Pharmacol, 1999, 55, 8–13. [Google Scholar] [CrossRef]
- Shen, M. , et al., Cannabinoid receptor agonists inhibit glutamatergic synaptic transmission in rat hippocampal cultures. J Neurosci, 1996, 16, 4322–4334. [Google Scholar]
- Wright, W.J., O. M. Schluter, and Y. Dong, A Feedforward Inhibitory Circuit Mediated by CB1-Expressing Fast-Spiking Interneurons in the Nucleus Accumbens. Neuropsychopharmacology, 2017, 42, 1146–1156. [Google Scholar]
- Robbe, D. , et al., Localization and mechanisms of action of cannabinoid receptors at the glutamatergic synapses of the mouse nucleus accumbens. J Neurosci, 2001, 21, 109–116. [Google Scholar]
- Robbe, D. , et al., Endogenous cannabinoids mediate long-term synaptic depression in the nucleus accumbens. Proc Natl Acad Sci U S A, 2002, 99, 8384–8388. [Google Scholar]
- Zlebnik, N.E. and J.F. Cheer, Drug-Induced Alterations of Endocannabinoid-Mediated Plasticity in Brain Reward Regions. J Neurosci, 2016, 36, 10230–10238. [Google Scholar] [CrossRef]
- Novak, M. , et al., Incentive learning underlying cocaine-seeking requires mGluR5 receptors located on dopamine D1 receptor-expressing neurons. J Neurosci, 2010, 30, 11973–11982. [Google Scholar]
- Fusco, F.R. , et al., Immunolocalization of CB1 receptor in rat striatal neurons: a confocal microscopy study. Synapse, 2004, 53, 159–167. [Google Scholar] [PubMed]
- Basavarajappa, B.S. , et al., Endocannabinoid system in neurodegenerative disorders. J Neurochem, 2017, 142, 624–648. [Google Scholar] [PubMed]
- Ferrari, R. , et al., FTD and ALS: a tale of two diseases. Curr Alzheimer Res 2011, 8, 273–294. [Google Scholar]
- Gibson, S.B. , et al., Familial clustering of ALS in a population-based resource. Neurology, 2014, 82, 17–22. [Google Scholar]
- Martin, S. Al Khleifat, and A. Al-Chalabi, What causes amyotrophic lateral sclerosis? F1000Res, 2017, 6, 371. [Google Scholar] [CrossRef]
- Hely, M.A. , et al., Sydney Multicenter Study of Parkinson's disease: non-L-dopa-responsive problems dominate at 15 years. Mov Disord, 2005, 20, 190–199. [Google Scholar]
- Reid, W.G. , et al., Dementia in Parkinson's disease: a 20-year neuropsychological study (Sydney Multicentre Study). J Neurol Neurosurg Psychiatry, 2011, 82, 1033–1037. [Google Scholar]
- Fernandez-Ruiz, J. , et al., Ageing, Neurodegeneration and the Endocannabinoid System. Curr Top Behav Neurosci 2025. [Google Scholar]
- Meanti, R. , et al., Cannabinoid Receptor 2 (CB2R) as potential target for the pharmacological treatment of neurodegenerative diseases. Biomed Pharmacother, 2025, 186, 118044. [Google Scholar]
- Oh, T.K. and I.A. Song, Impact of prescribed opioid use on development of dementia among patients with chronic non-cancer pain. Sci Rep, 2024, 14, 3313. [Google Scholar] [CrossRef] [PubMed]
- Beheshti, I. , Cocaine Destroys Gray Matter Brain Cells and Accelerates Brain Aging. Biology 2023, 12. [Google Scholar] [CrossRef]
- Rebolledo-Perez, L. , et al., Substance Abuse and Cognitive Decline: The Critical Role of Tau Protein as a Potential Biomarker. Int J Mol Sci 2025, 26. [Google Scholar] [CrossRef]
- Peng, B. , et al., Role of Alcohol Drinking in Alzheimer's Disease, Parkinson's Disease, and Amyotrophic Lateral Sclerosis. Int J Mol Sci 2020, 21. [Google Scholar] [CrossRef]
- Sabia, S. , et al., Alcohol consumption and risk of dementia: 23 year follow-up of Whitehall II cohort study. BMJ, 2018, 362, k2927. [Google Scholar] [PubMed]
- Koch, M. , et al., Alcohol Consumption and Risk of Dementia and Cognitive Decline Among Older Adults With or Without Mild Cognitive Impairment. JAMA Netw Open, 2019, 2, e1910319. [Google Scholar]
- Beaudet, G. , et al., Long-Lasting Effects of Chronic Intermittent Alcohol Exposure in Adolescent Mice on Object Recognition and Hippocampal Neuronal Activity. Alcohol Clin Exp Res, 2016, 40, 2591–2603. [Google Scholar]
- Vidal-Infer, A. , et al., Effect of intermittent exposure to ethanol and MDMA during adolescence on learning and memory in adult mice. Behav Brain Funct, 2012, 8, 32. [Google Scholar]
- Coleman, L.G., Jr. , et al., Adolescent binge drinking alters adult brain neurotransmitter gene expression, behavior, brain regional volumes, and neurochemistry in mice. Alcohol Clin Exp Res, 2011, 35, 671–688. [Google Scholar]
- Rico-Barrio, I. , et al., Cognitive and neurobehavioral benefits of an enriched environment on young adult mice after chronic ethanol consumption during adolescence. Addict Biol, 2019, 24, 969–980. [Google Scholar]
- Montagud-Romero, S. , et al., The novelty-seeking phenotype modulates the long-lasting effects of intermittent ethanol administration during adolescence. PLoS One, 2014, 9, e92576. [Google Scholar]
- Guerri, C. and M.Pascual, Mechanisms involved in the neurotoxic, cognitive, and neurobehavioral effects of alcohol consumption during adolescence. Alcohol 2010, 44, 15–26. [Google Scholar] [CrossRef] [PubMed]
- Alfonso-Loeches, S. and C.Guerri, Molecular and behavioral aspects of the actions of alcohol on the adult and developing brain. Crit Rev Clin Lab Sci 2011, 48, 19–47. [Google Scholar] [CrossRef]
- Espeland, M.A. , et al., Association between reported alcohol intake and cognition: results from the Women's Health Initiative Memory Study. Am J Epidemiol, 2005, 161, 228–238. [Google Scholar]
- Ganguli, M. , et al., Alcohol consumption and cognitive function in late life: a longitudinal community study. Neurology, 2005, 65, 1210–1217. [Google Scholar]
- Stampfer, M.J. , et al., Effects of moderate alcohol consumption on cognitive function in women. N Engl J Med, 2005, 352, 245–253. [Google Scholar]
- Stott, D.J. , et al., Does low to moderate alcohol intake protect against cognitive decline in older people? J Am Geriatr Soc, 2008, 56, 2217–2224. [Google Scholar] [CrossRef] [PubMed]
- Lobo, E. , et al., Is there an association between low-to-moderate alcohol consumption and risk of cognitive decline? Am J Epidemiol, 2010, 172, 708–716. [Google Scholar] [CrossRef] [PubMed]
- Yen, F.S. , et al., The Impact of Alcohol Consumption on Cognitive Impairment in Patients With Diabetes, Hypertension, or Chronic Kidney Disease. Front Med (Lausanne), 2022, 9, 861145. [Google Scholar]
- Kabai, P. , Alcohol consumption and cognitive decline in early old age. Neurology, 2014, 83, 476. [Google Scholar] [CrossRef]
- Kim, J.W. , et al., Association of moderate alcohol intake with in vivo amyloid-beta deposition in human brain: A cross-sectional study. PLoS Med, 2020, 17, e1003022. [Google Scholar]
- Flanigan, M.R. , et al., Imaging beta-amyloid (Abeta) burden in the brains of middle-aged individuals with alcohol-use disorders: a [(11)C]PIB PET study. Transl Psychiatry, 2021, 11, 257. [Google Scholar]
- Mukamal, K.J. , et al., Prospective study of alcohol consumption and risk of dementia in older adults. JAMA, 2003, 289, 1405–1413. [Google Scholar]
- Harwood, D.G. , et al., The effect of alcohol and tobacco consumption, and apolipoprotein E genotype, on the age of onset in Alzheimer's disease. Int J Geriatr Psychiatry, 2010, 25, 511–518. [Google Scholar]
- Palacios, N. , et al., Alcohol and risk of Parkinson's disease in a large, prospective cohort of men and women. Mov Disord, 2012, 27, 980–987. [Google Scholar]
- Eriksson, A.K. , et al., Alcohol use disorders and risk of Parkinson's disease: findings from a Swedish national cohort study 1972-2008. BMC Neurol, 2013, 13, 190. [Google Scholar]
- Rotermund, C. , et al., Enhanced motivation to alcohol in transgenic mice expressing human alpha-synuclein. J Neurochem, 2017, 143, 294–305. [Google Scholar]
- D'Ovidio, F. , et al., Association between alcohol exposure and the risk of amyotrophic lateral sclerosis in the Euro-MOTOR study. J Neurol Neurosurg Psychiatry, 2019, 90, 11–19. [Google Scholar]
- Mesquita, J. Silva, and A.Machado, Delayed Huntington's disease diagnosis in two alcoholic patients with a family history of "Parkinson's disease". J Neuropsychiatry Clin Neurosci, 2010, 22, 451–e2. [Google Scholar] [CrossRef]
- Basavarajappa, B.S. , Endocannabinoid System and Alcohol Abuse Disorders. Adv Exp Med Biol, 2019, 1162, 89–127. [Google Scholar]
- Basavarajappa, B.S. , et al., Distinct functions of endogenous cannabinoid system in alcohol abuse disorders. Br J Pharmacol, 2019, 176, 3085–3109. [Google Scholar]
- Serrano, A. and L.A.Natividad, Alcohol-Endocannabinoid Interactions: Implications for Addiction-Related Behavioral Processes. Alcohol Res, 2022, 42, 09. [Google Scholar] [CrossRef]
- Wolfe, S.A., V. Vozella, and M. Roberto, The Synaptic Interactions of Alcohol and the Endogenous Cannabinoid System. Alcohol Res, 2022, 42, 03. [Google Scholar] [PubMed]
- Serrano, A. , et al., Deficient endocannabinoid signaling in the central amygdala contributes to alcohol dependence-related anxiety-like behavior and excessive alcohol intake. Neuropsychopharmacology, 2018, 43, 1840–1850. [Google Scholar]
- Rivera, P. , et al., Pharmacological activation of CB2 receptors counteracts the deleterious effect of ethanol on cell proliferation in the main neurogenic zones of the adult rat brain. Front Cell Neurosci, 2015, 9, 379. [Google Scholar] [PubMed]
- Bellozi, P.M.Q. , et al., URB597 ameliorates the deleterious effects induced by binge alcohol consumption in adolescent rats. Neurosci Lett, 2019, 711, 134408. [Google Scholar] [PubMed]
- Sanchez-Marin, L. , et al., Effects of Intermittent Alcohol Exposure on Emotion and Cognition: A Potential Role for the Endogenous Cannabinoid System and Neuroinflammation. Front Behav Neurosci, 2017, 11, 15. [Google Scholar]
- Erdozain, A.M. and L.F.Callado, Involvement of the endocannabinoid system in alcohol dependence: the biochemical, behavioral and genetic evidence. Drug Alcohol Depend, 2011, 117, 102–110. [Google Scholar] [CrossRef] [PubMed]
- Subbanna, S. , et al., Anandamide-CB1 receptor signaling contributes to postnatal ethanol-induced neonatal neurodegeneration, adult synaptic, and memory deficits. J Neurosci, 2013, 33, 6350–6366. [Google Scholar]
- Subbanna, S. , et al., Postnatal ethanol exposure alters levels of 2-arachidonylglycerol-metabolizing enzymes and pharmacological inhibition of monoacylglycerol lipase does not cause neurodegeneration in neonatal mice. J Neurochem, 2015, 134, 276–287. [Google Scholar]
- Subbanna, S. , et al., Ethanol exposure induces neonatal neurodegeneration by enhancing CB1R Exon1 histone H4K8 acetylation and up-regulating CB1R function causing neurobehavioral abnormalities in adult mice. Int J Neuropsychopharmacol 2014, 18. [Google Scholar]
- Subbanna, S. , et al., CB1R-Mediated Activation of Caspase-3 Causes Epigenetic and Neurobehavioral Abnormalities in Postnatal Ethanol-Exposed Mice. Front Mol Neurosci, 2018, 11, 45. [Google Scholar] [PubMed]
- Subbanna, S. and B.S. Basavarajappa, Postnatal Ethanol-Induced Neurodegeneration Involves CB1R-Mediated beta-Catenin Degradation in Neonatal Mice. Brain Sci 2020, 10. [Google Scholar] [CrossRef]
- Nagre, N.N. , et al., CB1-receptor knockout neonatal mice are protected against ethanol-induced impairments of DNMT1, DNMT3A, and DNA methylation. J Neurochem, 2015, 132, 429–442. [Google Scholar] [CrossRef]
- Joshi, V. , et al., CB1R regulates CDK5 signaling and epigenetically controls Rac1 expression contributing to neurobehavioral abnormalities in mice postnatally exposed to ethanol. Neuropsychopharmacology 2019, 44, 514–525. [Google Scholar] [CrossRef] [PubMed]
- Basavarajappa, B.S. , et al., Elevation of endogenous anandamide impairs LTP, learning, and memory through CB1 receptor signaling in mice. Hippocampus 2014, 24, 808–818. [Google Scholar] [CrossRef]
- Ledesma, J.C. , et al., Adolescent binge-ethanol accelerates cognitive impairment and beta-amyloid production and dysregulates endocannabinoid signaling in the hippocampus of APP/PSE mice. Addict Biol 2021, 26, e12883. [Google Scholar] [CrossRef]
- Santos-Garcia, I. , et al., Preclinical investigation in FAAH inhibition as a neuroprotective therapy for frontotemporal dementia using TDP-43 transgenic male mice. J Neuroinflammation, 2023, 20, 108. [Google Scholar] [CrossRef]
- Arnanz, M.A. , et al., Fatty acid amide hydrolase gene inactivation induces hetero-cellular potentiation of microglial function in the 5xFAD mouse model of Alzheimer's disease. Glia, 2025, 73, 352–367. [Google Scholar] [CrossRef] [PubMed]
- Armeli, F. , et al., FAAH Inhibition Counteracts Neuroinflammation via Autophagy Recovery in AD Models. Int J Mol Sci 2024, 25. [Google Scholar] [CrossRef] [PubMed]
- Galan-Ganga, M. , et al., Cannabinoid receptor CB2 ablation protects against TAU induced neurodegeneration. Acta Neuropathol Commun 2021, 9, 90. [Google Scholar] [CrossRef]
- Clark, K.H. A.Wiley, and C.W. Bradberry, Psychostimulant abuse and neuroinflammation: emerging evidence of their interconnection. Neurotox Res 2013, 23, 174–188. [Google Scholar] [CrossRef]
- Pereira, R.B. B.Andrade, and P. Valentao, A Comprehensive View of the Neurotoxicity Mechanisms of Cocaine and Ethanol. Neurotox Res 2015, 28, 253–267. [Google Scholar] [CrossRef]
- Liu, S. , et al., Disrupted autophagy after spinal cord injury is associated with ER stress and neuronal cell death. Cell Death Dis 2015, 6, e1582. [Google Scholar] [CrossRef] [PubMed]
- Doyle, K.M. , et al., Unfolded proteins and endoplasmic reticulum stress in neurodegenerative disorders. J Cell Mol Med 2011, 15, 2025–2039. [Google Scholar] [CrossRef]
- Costa, B.M. , et al., Role of endoplasmic reticulum (ER) stress in cocaine-induced microglial cell death. J Neuroimmune Pharmacol 2013, 8, 705–714. [Google Scholar] [CrossRef]
- Periyasamy, P. L. Guo, and S.Buch, Cocaine induces astrocytosis through ER stress-mediated activation of autophagy. Autophagy 2016, 12, 1310–1329. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.J. , et al., Alzheimer-like phosphorylation of tau and neurofilament induced by cocaine in vivo. Acta Pharmacol Sin 2003, 24, 512–518. [Google Scholar]
- Arendt, R.E. Minnes, and L.T.Singer, Fetal Cocaine Exposure: Neurologic Effects and Sensory-Motor Delays. Phys Occup Ther Pediatr 1996, 16, 129–144. [Google Scholar] [CrossRef]
- Ferreira, C. , et al., Neuroprotection or Neurotoxicity of Illicit Drugs on Parkinson's Disease. Life (Basel) 2020, 10. [Google Scholar]
- Wen, S. , et al., Role of Mitochondrial Dynamics in Cocaine's Neurotoxicity. Int J Mol Sci 2022, 23. [Google Scholar] [CrossRef] [PubMed]
- Lee, F.J. , et al., Direct binding and functional coupling of alpha-synuclein to the dopamine transporters accelerate dopamine-induced apoptosis. FASEB J 2001, 15, 916–926. [Google Scholar]
- Qin, Y. , et al., Cocaine abuse elevates alpha-synuclein and dopamine transporter levels in the human striatum. Neuroreport 2005, 16, 1489–1493. [Google Scholar] [CrossRef] [PubMed]
- Bauer, L.O. , Psychomotor and electroencephalographic sequelae of cocaine dependence. NIDA Res Monogr, 1996, 163, 66–93. [Google Scholar]
- Argyriou, A.A. , et al., Cocaine use and abuse triggering sporadic young-onset amyotrophic lateral sclerosis. Neurodegener Dis, 2011, 8, 146–148. [Google Scholar] [CrossRef]
- Rockhold, R.W. , Glutamatergic involvement in psychomotor stimulant action. Prog Drug Res, 1998, 50, 155–192. [Google Scholar] [PubMed]
- Heath, P.R. and P.J. Shaw, Update on the glutamatergic neurotransmitter system and the role of excitotoxicity in amyotrophic lateral sclerosis. Muscle Nerve 2002, 26, 438–458. [Google Scholar] [CrossRef]
- Nguyen, X.K. , et al., Liposomal melatonin rescues methamphetamine-elicited mitochondrial burdens, pro-apoptosis, and dopaminergic degeneration through the inhibition PKCdelta gene. J Pineal Res, 2015, 58, 86–106. [Google Scholar] [CrossRef]
- Shukla, M. and B.Vincent, The multi-faceted impact of methamphetamine on Alzheimer's disease: From a triggering role to a possible therapeutic use. Ageing Res Rev, 2020, 60, 101062. [Google Scholar] [CrossRef]
- Granado, N. Ares-Santos, and R.Moratalla, Methamphetamine and Parkinson's disease. Parkinsons Dis, 2013, 2013, 308052. [Google Scholar] [PubMed]
- Ares-Santos, S. Granado, and R.Moratalla, The role of dopamine receptors in the neurotoxicity of methamphetamine. J Intern Med 2013, 273, 437–453. [Google Scholar] [CrossRef]
- Ares-Santos, S. , et al., Methamphetamine causes degeneration of dopamine cell bodies and terminals of the nigrostriatal pathway evidenced by silver staining. Neuropsychopharmacology 2014, 39, 1066–1080. [Google Scholar] [CrossRef]
- Chen, L. , et al., Methamphetamine exposure upregulates the amyloid precursor protein and hyperphosphorylated tau expression: The roles of insulin signaling in SH-SY5Y cell line. J Toxicol Sci 2019, 44, 493–503. [Google Scholar] [CrossRef]
- Callaghan, R.C. , et al., Increased risk of Parkinson's disease in individuals hospitalized with conditions related to the use of methamphetamine or other amphetamine-type drugs. Drug Alcohol Depend 2012, 120, 35–40. [Google Scholar] [CrossRef] [PubMed]
- Biagioni, F. , et al., Methamphetamine persistently increases alpha-synuclein and suppresses gene promoter methylation within striatal neurons. Brain Res 2019, 1719, 157–175. [Google Scholar] [CrossRef]
- Fernandez-Ruiz, J. A.Moro, and J. Martinez-Orgado, Cannabinoids in Neurodegenerative Disorders and Stroke/Brain Trauma: From Preclinical Models to Clinical Applications. Neurotherapeutics 2015, 12, 793–806. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Ruiz, J. Romero, and J.A. Ramos, Endocannabinoids and Neurodegenerative Disorders: Parkinson's Disease, Huntington's Chorea, Alzheimer's Disease, and Others. Handb Exp Pharmacol 2015, 231, 233–259. [Google Scholar]
- Marsicano, G. , et al., CB1 cannabinoid receptors and on-demand defense against excitotoxicity. Science 2003, 302, 84–88. [Google Scholar] [CrossRef]
- Fernandez-Moncada, I. , et al., Type-1 cannabinoid receptors and their ever-expanding roles in brain energy processes. J Neurochem 2024, 168, 693–703. [Google Scholar] [CrossRef] [PubMed]
- Rozenfeld, R. and L.A.Devi, Regulation of CB1 cannabinoid receptor trafficking by the adaptor protein AP-3. FASEB J, 2008, 22, 2311–2322. [Google Scholar] [CrossRef]
- Brailoiu, G.C. , et al., Intracellular cannabinoid type 1 (CB1) receptors are activated by anandamide. J Biol Chem, 2011, 286, 29166–29174. [Google Scholar] [CrossRef] [PubMed]
- Arevalo-Martin, A. , et al., CB2 cannabinoid receptors as an emerging target for demyelinating diseases: from neuroimmune interactions to cell replacement strategies. Br J Pharmacol, 2008, 153, 216–225. [Google Scholar] [CrossRef]
- Duarte, J.M. , et al., CB(1) receptor activation inhibits neuronal and astrocytic intermediary metabolism in the rat hippocampus. Neurochem Int, 2012, 60, 1–8. [Google Scholar] [CrossRef]
- Molina-Holgado, F. , et al., Endogenous interleukin-1 receptor antagonist mediates anti-inflammatory and neuroprotective actions of cannabinoids in neurons and glia. J Neurosci 2003, 23, 6470–6474. [Google Scholar] [CrossRef]
- Stella, N. , Cannabinoid and cannabinoid-like receptors in microglia, astrocytes, and astrocytomas. Glia, 2010, 58, 1017–1030. [Google Scholar] [CrossRef]
- Benito, C. , et al., Cannabinoid CB2 receptors and fatty acid amide hydrolase are selectively overexpressed in neuritic plaque-associated glia in Alzheimer's disease brains. J Neurosci, 2003, 23, 11136–11141. [Google Scholar] [CrossRef] [PubMed]
- Cabral, G.A. and L. Griffin-Thomas, Emerging role of the cannabinoid receptor CB2 in immune regulation: therapeutic prospects for neuroinflammation. Expert Rev Mol Med, 2009, 11, e3. [Google Scholar] [CrossRef]
- Cassano, T. , et al., Cannabinoid Receptor 2 Signaling in Neurodegenerative Disorders: From Pathogenesis to a Promising Therapeutic Target. Front Neurosci 2017, 11, 30. [Google Scholar] [CrossRef]
- Zoppi, S. , et al., Regulatory role of the cannabinoid CB2 receptor in stress-induced neuroinflammation in mice. Br J Pharmacol, 2014, 171, 2814–2826. [Google Scholar] [CrossRef]
- Choi, I.Y. , et al., Activation of cannabinoid CB2 receptor-mediated AMPK/CREB pathway reduces cerebral ischemic injury. Am J Pathol 2013, 182, 928–939. [Google Scholar] [CrossRef]
- Mechoulam, R. and E.Shohami, Endocannabinoids and traumatic brain injury. Mol Neurobiol, 2007, 36, 68–74. [Google Scholar] [CrossRef]
- Vendel, E. and E.C. de Lange, Functions of the CB1 and CB 2 receptors in neuroprotection at the level of the blood-brain barrier. Neuromolecular Med 2014, 16, 620–642. [Google Scholar] [CrossRef] [PubMed]
- Haj-Dahmane, S. and R.Y. Shen, Modulation of the serotonin system by endocannabinoid signaling. Neuropharmacology 2011, 61, 414–420. [Google Scholar] [CrossRef]
- Fujii, M. , et al., Cannabinoid type 2 receptor stimulation attenuates brain edema by reducing cerebral leukocyte infiltration following subarachnoid hemorrhage in rats. J Neurol Sci, 2014, 342, 101–106. [Google Scholar] [CrossRef] [PubMed]
- Hagan, K. Varelas, and H.Zheng, Endocannabinoid System of the Blood-Brain Barrier: Current Understandings and Therapeutic Potentials. Cannabis Cannabinoid Res, 2022, 7, 561–568. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Schematic representation of CB1R signaling pathways. Both endogenous and mock cannabinoids exert their effects primarily through activation of CB1R [15,16,17,18]. CB1Rs are seven-transmembrane GPCRs embedded in the neuronal cell membrane [19]. Upon activation, CB1Rs inhibit AC activity [20,21,22], suppress N-type and P/Q-type voltage-gated calcium channels [23,24,25,26], and modulate potassium channels by inhibiting D-type and activating A-type as well as GIRKs [23,24,25,26,27]. CB1R signaling reduces presynaptic calcium influx through these mechanisms, thereby regulating neurotransmitter release and inhibiting synaptic transmission [28,29]. The Gα subunit primarily mediates the inhibitory actions on AC and calcium channels. At the same time, the Gβγ complex activates GIRKs and PI3K. Subsequent activation of the p38, JNK, and ERK1/2 pathways leads to phosphorylation of downstream effectors such as cPLA2, ELK-1, c-Fos, c-Jun, and CREB, promoting the transcription of target genes including krox-24 and BDNF [16,30]. PI3K also mediates AKT-dependent inhibition of CREB activation [30]. Furthermore, CB1R-induced inhibition of AC decreases cAMP levels, thereby reducing protein kinase A (PKA) activity and the phosphorylation of specific K⁺ channels [18,31,32]. Under certain physiological conditions, CB1R signaling can also suppress ERK1/2 activation, leading to reduced CaMKIV and CREB phosphorylation, and consequent inhibition of Arc gene expression [33,34]. Stimulatory effects are indicated by arrows (→) and inhibitory effects by blunt-ended lines (⊥). CB1R, cannabinoid receptor 1; GPCRs, G protein–coupled receptors; AC, adenylate cyclase; GIRKs, G protein–coupled inwardly rectifying potassium channels; PI3K, phos phoinositide 3-kinase; p38, p38 mitogen-activated protein kinase; JNK, Jun N-terminal kinase; ERK, extracellular signal-regulated kinases, cPLA2, cytosolic phospholipase A2; Elk-1, E26 transformation-specific domain containing protein 1; c-Fos, an inducible transcription factor that regulates the delayed onset of effector genes; c-Jun, stress-activated protein; CREB, cAMP-response element binding protein; AKT, protein kinase B; cAMP, cyclic adenosine monophosphate; CaMKIV, calcium/calmodulin-dependent protein kinase IV; PKA protein kinase A. (Figure was created with BioRender.com).
Figure 1.
Schematic representation of CB1R signaling pathways. Both endogenous and mock cannabinoids exert their effects primarily through activation of CB1R [15,16,17,18]. CB1Rs are seven-transmembrane GPCRs embedded in the neuronal cell membrane [19]. Upon activation, CB1Rs inhibit AC activity [20,21,22], suppress N-type and P/Q-type voltage-gated calcium channels [23,24,25,26], and modulate potassium channels by inhibiting D-type and activating A-type as well as GIRKs [23,24,25,26,27]. CB1R signaling reduces presynaptic calcium influx through these mechanisms, thereby regulating neurotransmitter release and inhibiting synaptic transmission [28,29]. The Gα subunit primarily mediates the inhibitory actions on AC and calcium channels. At the same time, the Gβγ complex activates GIRKs and PI3K. Subsequent activation of the p38, JNK, and ERK1/2 pathways leads to phosphorylation of downstream effectors such as cPLA2, ELK-1, c-Fos, c-Jun, and CREB, promoting the transcription of target genes including krox-24 and BDNF [16,30]. PI3K also mediates AKT-dependent inhibition of CREB activation [30]. Furthermore, CB1R-induced inhibition of AC decreases cAMP levels, thereby reducing protein kinase A (PKA) activity and the phosphorylation of specific K⁺ channels [18,31,32]. Under certain physiological conditions, CB1R signaling can also suppress ERK1/2 activation, leading to reduced CaMKIV and CREB phosphorylation, and consequent inhibition of Arc gene expression [33,34]. Stimulatory effects are indicated by arrows (→) and inhibitory effects by blunt-ended lines (⊥). CB1R, cannabinoid receptor 1; GPCRs, G protein–coupled receptors; AC, adenylate cyclase; GIRKs, G protein–coupled inwardly rectifying potassium channels; PI3K, phos phoinositide 3-kinase; p38, p38 mitogen-activated protein kinase; JNK, Jun N-terminal kinase; ERK, extracellular signal-regulated kinases, cPLA2, cytosolic phospholipase A2; Elk-1, E26 transformation-specific domain containing protein 1; c-Fos, an inducible transcription factor that regulates the delayed onset of effector genes; c-Jun, stress-activated protein; CREB, cAMP-response element binding protein; AKT, protein kinase B; cAMP, cyclic adenosine monophosphate; CaMKIV, calcium/calmodulin-dependent protein kinase IV; PKA protein kinase A. (Figure was created with BioRender.com).
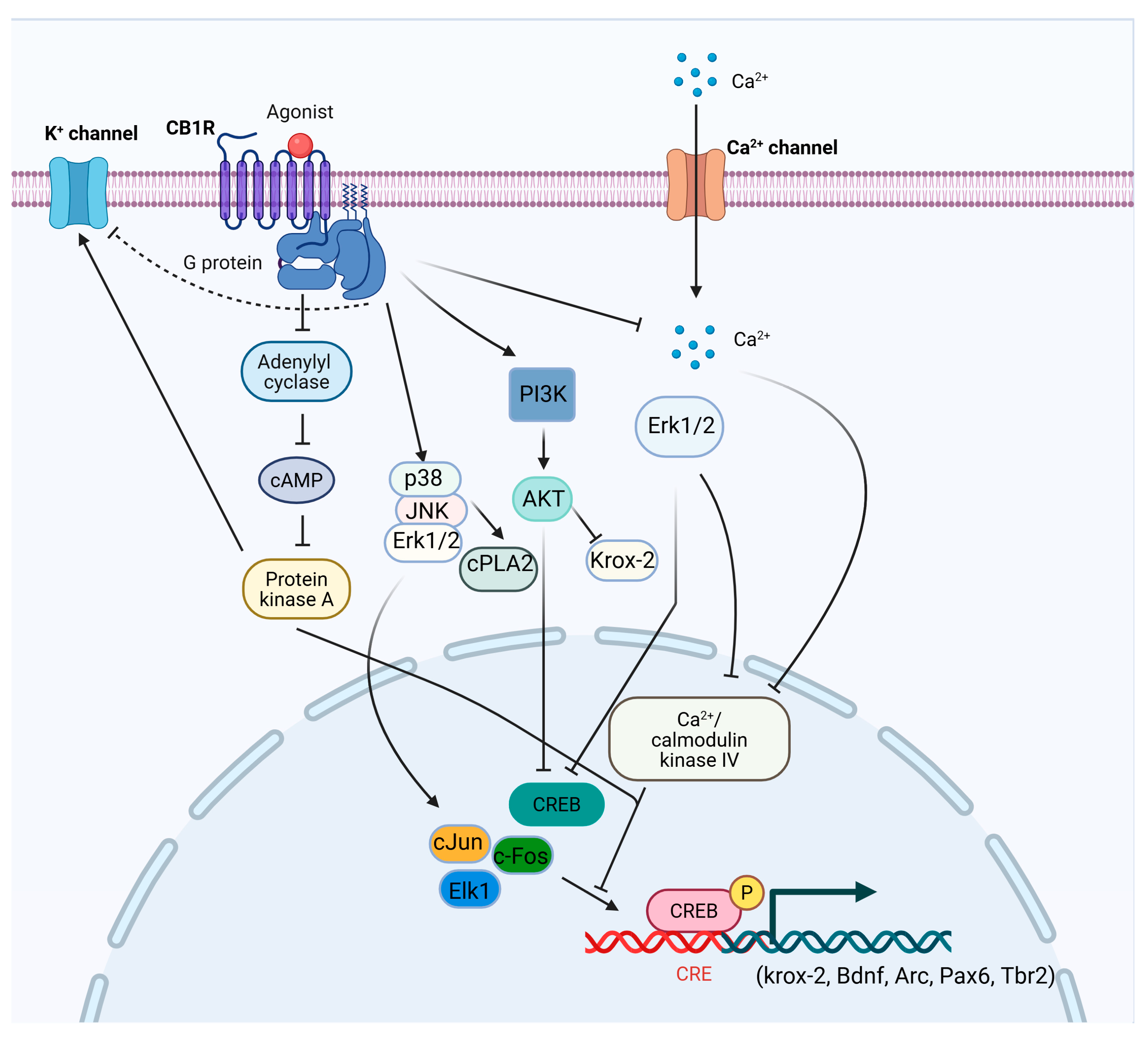
Figure 2.
Schematic representation of CB2R signaling pathways. Both endogenous and synthetic cannabinoids interact with CB2R, a seven-transmembrane GPCR localized predominantly on immune cells, microglia, and, to a lesser extent, neurons [62,65,82,83]. Upon activation, CB2Rs couple primarily to Gi/o proteins, leading to inhibition of AC and a consequent reduction in intracellular cAMP levels. This attenuates PKA activity, thereby modulating downstream phosphorylation events that regulate cellular excitability, immune signaling, and gene transcription. CB2R activation also recruits multiple intracellular signaling cascades, including the MAPK pathways—ERK1/2 and p38 MAPK, and the PI3K/AKT pathway. These pathways contribute to cell survival, proliferation, and differentiation, and play critical roles in regulating neuroinflammation and neuroprotection. In immune and glial cells, CB2R-mediated signaling can modulate cytokine production, inhibit pro-inflammatory responses, and promote anti-inflammatory or restorative phenotypes. Additionally, CB2R activation has been linked to the de novo synthesis of ceramide, a bioactive lipid that serves as a second messenger in pathways governing apoptosis, oxidative stress responses, and cell cycle arrest [62,75,84,85,86,87,88,89,90,91]. CB2R signaling exerts broad regulatory effects on immune and neuronal function through these combined mechanisms. Stimulatory effects are indicated by arrows (→) and inhibitory effects by blunt-ended lines (⊥). CB2R, Cannabinoid Receptor 2; GPCR, G protein–coupled receptor; AC, adenylate cyclase; PKA, protein kinase A; MAPK, mitogen-activated protein kinase; ERK, extracellular signal-regulated kinases; p38 MAPK, p38 mitogen-activated protein kinase; PI3K, phosphoinositide 3-kinase; AKT, protein kinase B.
Figure 2.
Schematic representation of CB2R signaling pathways. Both endogenous and synthetic cannabinoids interact with CB2R, a seven-transmembrane GPCR localized predominantly on immune cells, microglia, and, to a lesser extent, neurons [62,65,82,83]. Upon activation, CB2Rs couple primarily to Gi/o proteins, leading to inhibition of AC and a consequent reduction in intracellular cAMP levels. This attenuates PKA activity, thereby modulating downstream phosphorylation events that regulate cellular excitability, immune signaling, and gene transcription. CB2R activation also recruits multiple intracellular signaling cascades, including the MAPK pathways—ERK1/2 and p38 MAPK, and the PI3K/AKT pathway. These pathways contribute to cell survival, proliferation, and differentiation, and play critical roles in regulating neuroinflammation and neuroprotection. In immune and glial cells, CB2R-mediated signaling can modulate cytokine production, inhibit pro-inflammatory responses, and promote anti-inflammatory or restorative phenotypes. Additionally, CB2R activation has been linked to the de novo synthesis of ceramide, a bioactive lipid that serves as a second messenger in pathways governing apoptosis, oxidative stress responses, and cell cycle arrest [62,75,84,85,86,87,88,89,90,91]. CB2R signaling exerts broad regulatory effects on immune and neuronal function through these combined mechanisms. Stimulatory effects are indicated by arrows (→) and inhibitory effects by blunt-ended lines (⊥). CB2R, Cannabinoid Receptor 2; GPCR, G protein–coupled receptor; AC, adenylate cyclase; PKA, protein kinase A; MAPK, mitogen-activated protein kinase; ERK, extracellular signal-regulated kinases; p38 MAPK, p38 mitogen-activated protein kinase; PI3K, phosphoinositide 3-kinase; AKT, protein kinase B.
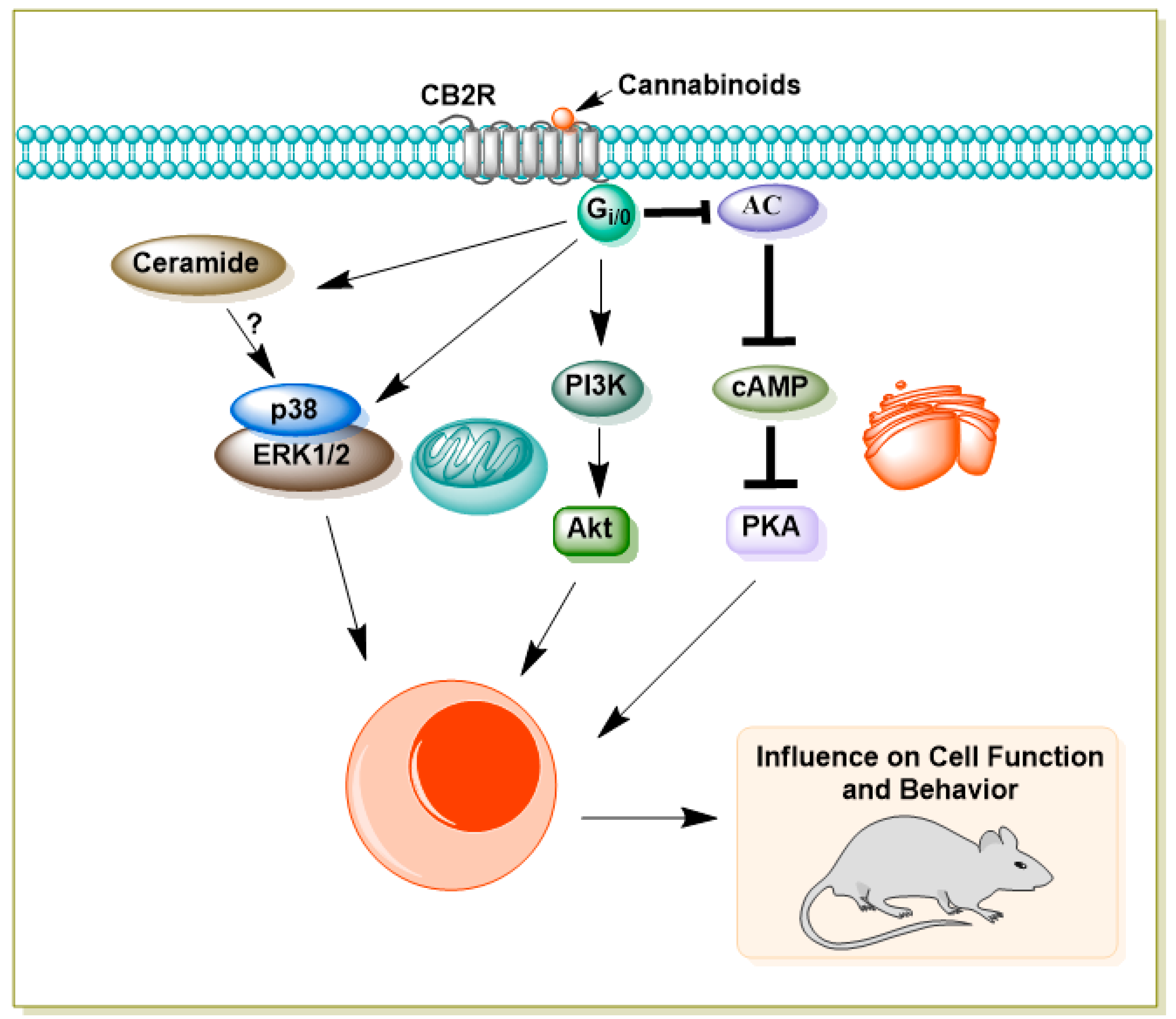
Figure 3.
Chemical structures of endogenous cannabinoids anandamide (AEA) and 2-arachidonoylglycerol (2-AG). The two principal endocannabinoids, AEA and 2-AG, are lipid-derived signaling molecules synthesized on demand from membrane phospholipid precursors. Both AEA and 2-AG act as endogenous ligands for cannabinoid receptors CB1R and CB2R, mediating a wide range of physiological and neurobiological processes.
Figure 3.
Chemical structures of endogenous cannabinoids anandamide (AEA) and 2-arachidonoylglycerol (2-AG). The two principal endocannabinoids, AEA and 2-AG, are lipid-derived signaling molecules synthesized on demand from membrane phospholipid precursors. Both AEA and 2-AG act as endogenous ligands for cannabinoid receptors CB1R and CB2R, mediating a wide range of physiological and neurobiological processes.
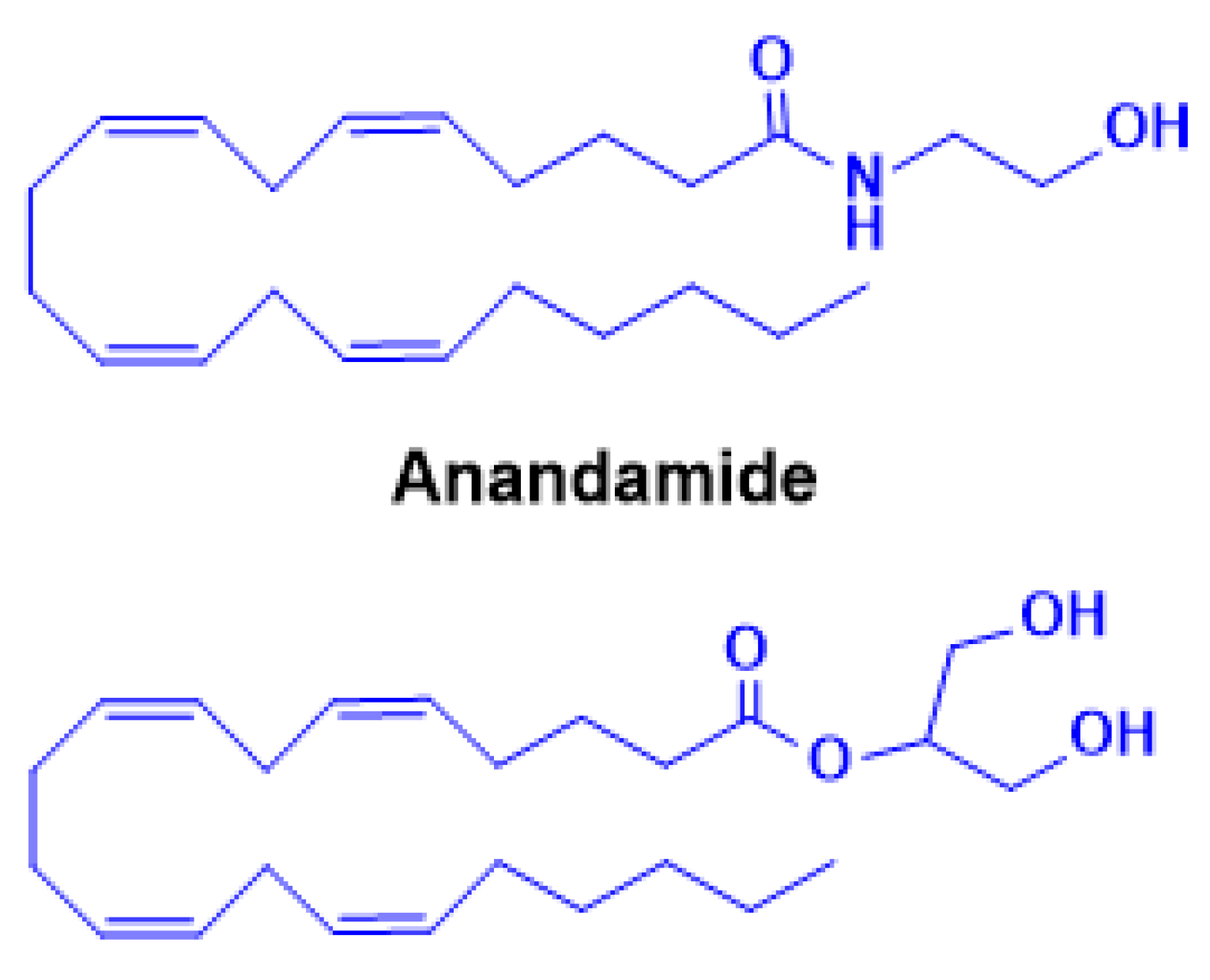
Figure 4.
Endocannabinoid-dependent mechanisms mediating natural reward and drug-seeking behaviors. Two major endocannabinoid (eCB)-dependent mechanisms within the mesocorticolimbic circuit contribute to both natural and drug-induced reward processing. (A) In VTA, eCB signaling regulates dopaminergic activity via disinhibition. Under basal conditions, DA neurons are tonically inhibited by GABAergic interneurons through GABAB receptor activation. Presentation of reward-predictive or drug-associated cues induces phasic DA firing, elevating intracellular Ca2+ and activating DAGL. DAGL catalyzes 2-AG synthesis, which acts retrogradely on presynaptic CB1Rs on GABA terminals, suppressing GABA release and disinhibiting DA neurons. This promotes burst firing and enhanced DA release [135,136,137]. Blockade of GABAB or CB1Rs prevents this reward-seeking response, confirming CB1R-dependent disinhibition as a key modulatory mechanism. (B) In the NAc, eCB–glutamate interactions fine-tune excitatory drive onto D1-MSNs. Glutamatergic inputs from the prefrontal cortex activate mGluR5, stimulating DAGL and 2-AG synthesis. The released 2-AG retrogradely activates CB1Rs on glutamatergic terminals, inhibiting further glutamate release and modulating excitatory transmission [138]. Inhibition of either mGluR5 or CB1Rs abolishes both natural and drug reward-seeking behaviors [139,140]. Reward-related glutamatergic afferents promote DA neuron burst firing via iGluR activation, balanced by GABAergic inhibition [141]. Reduced GABAergic tone disinhibits DA neurons, increasing DA release onto MSNs. Ca2+ influx through voltage-gated channels, together with mGluR1/5 activation, drives “on-demand” production of 2-AG (and to a lesser extent, AEA. Retrograde 2-AG signaling through CB1Rs on GABAergic and glutamatergic terminals—both MAGL—enhances phasic DA release and plasticity [113]. CB1Rs are denser on GABAergic than glutamatergic terminals, making cannabinoids (e.g., THC, 2-AG) full agonists at GABA sites but partial agonists at glutamatergic terminals [142,143,144]. The role of AEA remains less defined due to asymmetrical localization of its biosynthetic (NAPE-PLD) and degradative (FAAH) enzymes. CB1Rs on local GABAergic and glutamatergic terminals further regulate accumbal DA output—enhancing it via GABA inhibition or reducing it through glutamate suppression [145]. eCB, endocannabinoid; VTA, ventral tegmental area; GABAB, gamma-aminobutyric acid B; DAGL, diacylglycerol lipase; 2-AG, 2-arachidonoylglycerol; CB1Rs, cannabinoid type 1 receptors; NAc, nucleus accumbens; D1-MSNs, D1 medium spiny neurons; mGluR5, metabotropic lutamate receptor 5; iGluR, ionotropic glutamate receptor; NAPE, N-acyl-phosphatidylethanolamine; PLD, phospholipase D; FAAH, fatty acid amide hydrolasel; THC, tetrahydrocannabinol; GABA, γ-aminobutyric acid; ACh, acetylcholine. (Figure was created with BioRender.com).
Figure 4.
Endocannabinoid-dependent mechanisms mediating natural reward and drug-seeking behaviors. Two major endocannabinoid (eCB)-dependent mechanisms within the mesocorticolimbic circuit contribute to both natural and drug-induced reward processing. (A) In VTA, eCB signaling regulates dopaminergic activity via disinhibition. Under basal conditions, DA neurons are tonically inhibited by GABAergic interneurons through GABAB receptor activation. Presentation of reward-predictive or drug-associated cues induces phasic DA firing, elevating intracellular Ca2+ and activating DAGL. DAGL catalyzes 2-AG synthesis, which acts retrogradely on presynaptic CB1Rs on GABA terminals, suppressing GABA release and disinhibiting DA neurons. This promotes burst firing and enhanced DA release [135,136,137]. Blockade of GABAB or CB1Rs prevents this reward-seeking response, confirming CB1R-dependent disinhibition as a key modulatory mechanism. (B) In the NAc, eCB–glutamate interactions fine-tune excitatory drive onto D1-MSNs. Glutamatergic inputs from the prefrontal cortex activate mGluR5, stimulating DAGL and 2-AG synthesis. The released 2-AG retrogradely activates CB1Rs on glutamatergic terminals, inhibiting further glutamate release and modulating excitatory transmission [138]. Inhibition of either mGluR5 or CB1Rs abolishes both natural and drug reward-seeking behaviors [139,140]. Reward-related glutamatergic afferents promote DA neuron burst firing via iGluR activation, balanced by GABAergic inhibition [141]. Reduced GABAergic tone disinhibits DA neurons, increasing DA release onto MSNs. Ca2+ influx through voltage-gated channels, together with mGluR1/5 activation, drives “on-demand” production of 2-AG (and to a lesser extent, AEA. Retrograde 2-AG signaling through CB1Rs on GABAergic and glutamatergic terminals—both MAGL—enhances phasic DA release and plasticity [113]. CB1Rs are denser on GABAergic than glutamatergic terminals, making cannabinoids (e.g., THC, 2-AG) full agonists at GABA sites but partial agonists at glutamatergic terminals [142,143,144]. The role of AEA remains less defined due to asymmetrical localization of its biosynthetic (NAPE-PLD) and degradative (FAAH) enzymes. CB1Rs on local GABAergic and glutamatergic terminals further regulate accumbal DA output—enhancing it via GABA inhibition or reducing it through glutamate suppression [145]. eCB, endocannabinoid; VTA, ventral tegmental area; GABAB, gamma-aminobutyric acid B; DAGL, diacylglycerol lipase; 2-AG, 2-arachidonoylglycerol; CB1Rs, cannabinoid type 1 receptors; NAc, nucleus accumbens; D1-MSNs, D1 medium spiny neurons; mGluR5, metabotropic lutamate receptor 5; iGluR, ionotropic glutamate receptor; NAPE, N-acyl-phosphatidylethanolamine; PLD, phospholipase D; FAAH, fatty acid amide hydrolasel; THC, tetrahydrocannabinol; GABA, γ-aminobutyric acid; ACh, acetylcholine. (Figure was created with BioRender.com).
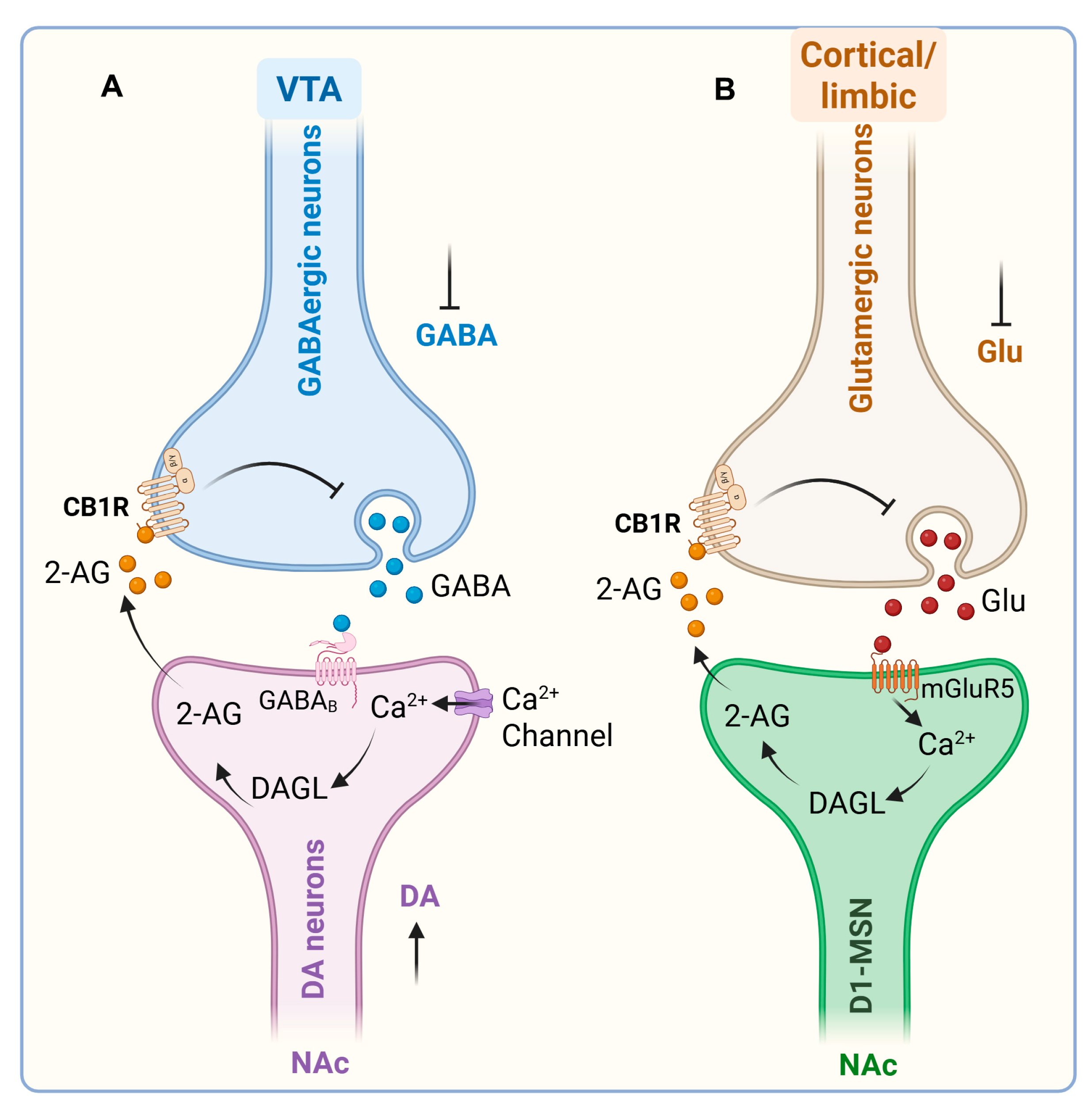
Figure 5.
Endocannabinoid system–mediated neuroprotection in neurodegenerative disorders. The eCBS provides neuroprotection through coordinated actions on neurons, astrocytes, microglia, and the vasculature. Neuronal protection involves CB1 receptor–mediated inhibition of excessive glutamate release at presynaptic terminals, modulation of postsynaptic calcium channels, and maintenance of glutamate/GABA balance, limiting excitotoxicity [151,234,235,236]. CB1 signaling also promotes autophagy, facilitating clearance of damaged proteins and aggregates [237,238,239]. Glial-mediated protection occurs via CB1, CB2, GPCRs, and PPAR receptor activation. In astrocytes, eCBs signaling supports energy metabolism, glutamate clearance, neurotrophin production, and anti-inflammatory factor release, enhancing neuronal homeostasis [157,234,235,237,240,241,242,243]. In microglia, CB2 activation promotes proliferation, migration to lesion sites, and M1-to-M2 phenotype conversion via NFκB inhibition, reducing neuroinflammation [243,244,245,246,247]. Vascular protection includes CB1/CB2-mediated maintenance of blood–brain barrier integrity, inhibition of leukocyte infiltration, β-amyloid clearance, and restoration of cerebral blood flow [248,249,250,251,252,253]. Collectively, eCBs signaling integrates neuronal, glial, and vascular mechanisms to preserve homeostasis, limit excitotoxicity and inflammation, and promote neuroprotection in ND disorders. ND, neurodegenerative; eCBS, endocannabinoid system; CB1R, cannabinoid type 1 receptor; GABA, gamma-aminobutyric Acid; GPCRs, G protein–coupled receptors; PPAR, proliferator-activated receptor; NFκB, nuclear factor kappa-light-chain-enhancer of activated B cells; CB1, cannabinoid receptors 1; CB2, cannabinoid receptors 2. (Figure was created with BioRender.com).
Figure 5.
Endocannabinoid system–mediated neuroprotection in neurodegenerative disorders. The eCBS provides neuroprotection through coordinated actions on neurons, astrocytes, microglia, and the vasculature. Neuronal protection involves CB1 receptor–mediated inhibition of excessive glutamate release at presynaptic terminals, modulation of postsynaptic calcium channels, and maintenance of glutamate/GABA balance, limiting excitotoxicity [151,234,235,236]. CB1 signaling also promotes autophagy, facilitating clearance of damaged proteins and aggregates [237,238,239]. Glial-mediated protection occurs via CB1, CB2, GPCRs, and PPAR receptor activation. In astrocytes, eCBs signaling supports energy metabolism, glutamate clearance, neurotrophin production, and anti-inflammatory factor release, enhancing neuronal homeostasis [157,234,235,237,240,241,242,243]. In microglia, CB2 activation promotes proliferation, migration to lesion sites, and M1-to-M2 phenotype conversion via NFκB inhibition, reducing neuroinflammation [243,244,245,246,247]. Vascular protection includes CB1/CB2-mediated maintenance of blood–brain barrier integrity, inhibition of leukocyte infiltration, β-amyloid clearance, and restoration of cerebral blood flow [248,249,250,251,252,253]. Collectively, eCBs signaling integrates neuronal, glial, and vascular mechanisms to preserve homeostasis, limit excitotoxicity and inflammation, and promote neuroprotection in ND disorders. ND, neurodegenerative; eCBS, endocannabinoid system; CB1R, cannabinoid type 1 receptor; GABA, gamma-aminobutyric Acid; GPCRs, G protein–coupled receptors; PPAR, proliferator-activated receptor; NFκB, nuclear factor kappa-light-chain-enhancer of activated B cells; CB1, cannabinoid receptors 1; CB2, cannabinoid receptors 2. (Figure was created with BioRender.com).
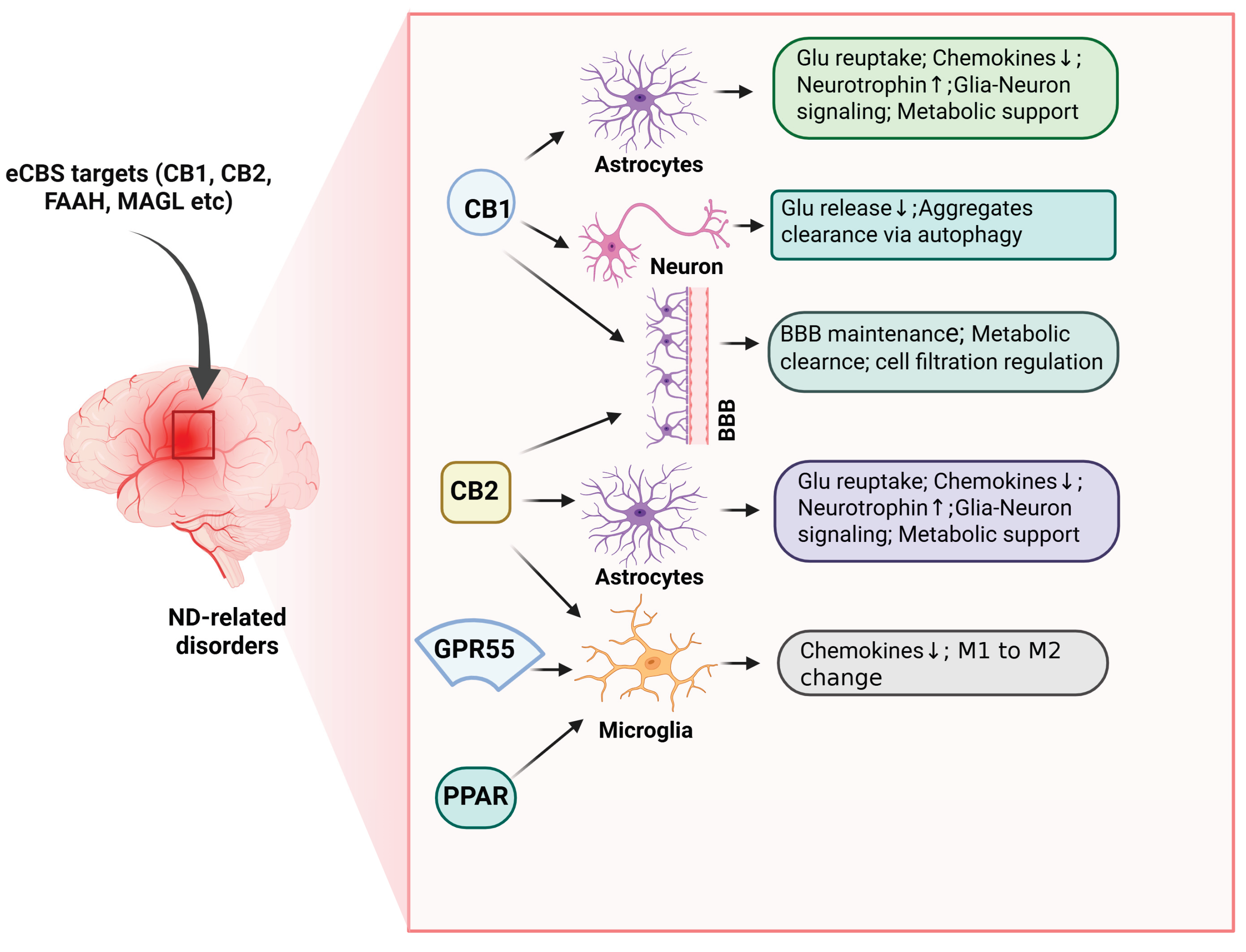
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



