Submitted:
20 August 2025
Posted:
21 August 2025
You are already at the latest version
Abstract
Schizophrenia is a severe, chronic mental disorder affecting near 1% of the global population, with about 60% of patients showing resistance to currently available antipsychotic drugs (APDs). The modern dopaminergic (DA) hypothesis of schizophrenia postulates that multiple pathogenic factors contribute to hyperdopaminergia in the striatum. Therefore, effective modulation of striatal DA remains a key strategy in the development of novel APDs. The striatum-enriched protein phosphatase (STEP) is selectively expressed in the dopaminergic neurons of the striatum together with D2 receptors - the main target of existing APDs. In this study, we investigated the APD-like effects of the STEP inhibitor TC-2153 in a genetic mouse model of schizophrenia (Disc1-L100P). We found that TC-2153 [10 mg/kg; i.p.] effectively reduced hyperactivity in Disc1-L100P mutant mice as assessed in the open field test. The STEP inhibitor also ameliorated disrupted latent inhibition (LI) in Disc1-L100P animals, but unexpectedly impaired LI in wild-type (WT) mice. In opposite, TC-2153 had no effect on deficient sensorimotor gating, measured by the pre-pulse inhibition (PPI) paradigm. Additionally, TC-2153 induced antidepressant-like effects in both WT and Disc1-L100P mutant mice, increasing their activity in the Forced Swim Test (FST). These new findings suggest that STEP inhibition produces APD-like effects in a genotype-specific manner while promoting antidepressant-related behavioural phenotypes regardless of genetic background. Overall, our preclinical results support STEP as a promising target for APD development and highlight a potential link between STEP and DISC1 molecular complex in the pathogenesis of schizophrenia – an avenue that warrants further investigation to better understand the molecular mechanisms underlying APD action.
Keywords:
DISC1
; STEP
; schizophrenia
; mouse model
; antipsychotic drug
; TC-2153
1. Introduction
Schizophrenia is a severe psychiatric disorder characterized by a disrupted sense of reality, which forms the core of its symptoms. This impairment significantly affects daily functioning and often leads to lifelong disability requiring continuous care. The dopaminergic (DA) hypothesis of schizophrenia was first proposed in 1976 [1] and has since evolved through the integration of new findings on the mechanisms of antipsychotic drugs (APDs) action and their effects on dopamine metabolism. A modern version of the DA hypothesis was introduced in 2009 [2] incorporating evidence from numerous pre-clinical and clinical studies that highlight the pathogenic role of gene-environment interactions in schizophrenia. For example, social isolation has been shown to enhance the pro-psychotic effects of psychostimulants [3] and unpredictable mild stress [4] on the DA system in rats. Similar observations have been reported in humans; increased stress-induced striatal DA release was reported in adults who experienced low maternal care during childhood [5]. Hence, the modern DA hypothesis of schizophrenia successfully combined multiple contributing factors that converge on a common pathway – elevated dopamine level in the striatum [2].
According to the World Health Organization (WHO), approximately 20 million people worldwide are affected by schizophrenia [6]. Despite the availability of APDs, their overall effectiveness remains limited, and they are often associated with a range of side effects. These include a high incident of movement disorders induced by atypical APDs, as well as metabolic syndromes such as weight gain, fatigue and hypersalivation. While APDs help stabilize schizophrenia symptoms over the long term and can facilitate remission, nearly 60% of patients show resistance to these treatments and frequently experience adverse effects. Dopamine D2 receptor (D2R) is the primary target for APDs development, and clinical effectiveness is closely linked to the drugs’ binding affinity to D2R [7]. D2R elicits their action via intracellular Gi/Go proteins, which control adenylate cyclase activity, phosphatidylinositol turnover, arachidonic acid release, ion channels activity (K+ and Ca2+), and protein kinases signaling [8]. Additionally, D2R can signal independently of G-proteins, for example through β-arrestin-2, which initiates the formation of a molecular complex involving protein phosphatase-2A (PP2A), protein kinase-B (Akt (PKB)) and glycogen synthase kinase-3 (GSK-3) [9]. Key mechanisms regulating D2R activity include kinase-dependent desensitization, receptor endocytosis, and endosomal trafficking, which facilitate complex formation with β-arrestin-2, adaptor protein-2, and clathrin [10].
Our study identified pathologically enhanced protein-protein interactions between D2R and DISC1 (Disrupted-In-Schizophrenia-1) in post-mortem brains of patients with schizophrenia and in the striatum of Disc1-L100P mutant mice - a genetic model of schizophrenia [11]. Notably, uncoupling the D2R-DISC1 interaction using a targeted peptide corrected schizophrenia-like behaviours in Disc1-L100P animals, suggesting antipsychotic potential. Furthermore, pharmacological inhibitors of GSK-3 [12] or PDE4 [12,13], both part of the DISC1 interactome, have shown APDs-like effects in pre-clinical studies. These findings underscore the critical role of DISC1 and its interactome in regulating dopamine function [14], offering promising new therapeutic targets for schizophrenia treatment.
STriatal-Enriched protein tyrosine Phosphatase (STEP; PTPN5) has been implicated in the pathophysiology of schizophrenia [15]. STEP is specifically expressed in dopaminergic neurons of the striatum [16], where D2Rs are also highly enriched, suggesting a potential role for STEP in modulating dopamine function. A study by Kim and colleagues [17] demonstrated a potential link between STEP and D2R interaction. So, genetic lack of D2R significantly reduced STEP levels in knockout mice, while genetic inhibition of STEP led to a decrease in dopaminergic neuron density. STEP substrates include key synaptic proteins such as the GluN2B subunit of NMDA receptors [18], the GluA2 subunit of AMPA receptors [19] and several kinases including ERK1/2, Fyn, and Pyk2 [20,21,22]. Functionally, STEP acts as a negative regulator of synaptic strength, opposing synaptic potentiation and plasticity [23].
The pharmacological STEP inhibitor, (8-(trifluoromethyl)benzo[f][1,2,3,4,5]pentathiepin-6-amine hydrochloride), TC-2153, is a benzopentathiepine compound containing five sulphur atoms. It was first discovered by our team as the first orally available synthetic pentathiepine [24] (Figure 1). A detailed description of the chemical synthesis of benzopentathiepine analogues and their biochemical characterization has been reported [25], and the behavioural effects of TC-2153 have been demonstrated in several recent studies. For instance, TC-2153 was able to reverse motor and cognitive deficits induced by PCP (an NMDA receptor blocker) in mice via the increased expression of brain-derived neurotrophic factor (BDNF) [26]. TC-2153 rescued short-term memory deficit in the Y-maze caused by the bacterial lipopolysaccharide exposure in mice, via increased phosphorylation of GluN2B, ERK1/2, CREB/BDNF, and PSD95 [27]. Beyond its cognitive effects, TC-2153 has also demonstrated antidepressant-like properties [28] and was found to ameliorate social deficit, anxiety and repetitive behaviour in a valproate-induced model of autism [29]. Recent studies have shown that TC-2153 mitigates fear-induced aggression in Norway rats selectively bred over 90 generations for heighten aggression toward humans. This behavioral improvement was accompanied by reduced expression of BDNF in the midbrain and increased expression of 5-HT1a receptors, along with elevated tryptophan hydroxylase activity in the hypothalamus [30,31,32].
STEP has emerged as a compelling molecular target for the treatment of mental disorders and may represent a promising candidate for the development of next-generation APDs, given its association with dopaminergic and NMDA systems as well as cognitive effects. Hence, we hypothesized that STEP may interact with DISC1 protein network and thereby exert APD-like effects. To test this hypothesis, we evaluated the APD-like effectiveness of the STEP inhibitor TC-2153 in Disc1-L100P mutant mice - a well-established genetic model of schizophrenia [34]. Our goal was to further support STEP as a new molecular target for APD development. In addition, we assessed the effects of TC-2153 in the Forced Swim Test, which is commonly used to evaluate depression-like behaviour in mice. This allowed us to expand upon previous findings [28] by examining TC-2153’s antidepressant potential in Disc1-L100P mice and comparing its APD-like capacity with its antidepressant-like actions.
2. Materials and Methods
2.1. Animals
Disc1-L100P homozygous male mice and their wild-type (WT) littermates, C57Bl/6NCrl (C57Bl/6N) inbred strain were bred in the animal facility of Scientific Research Institute of Neuroscience and Medicine (SRINM). Experiments were conducted on 2-3 months old Disc1-L100P\WT male mice. Homozygous Disc1-L100P and WT mice (DISC1-L100P+/+ or WT) were obtained from the breeding of heterozygous pairs. Genotyping was performed as described before [34], using primers: F, 5’-AGA CCA GGC TAC ATG AGA AGC-3’ and R: 5’-AAG CTG GAA GTG AAG GTG TCT-3’. All mice were housed 5 per cage in the vivarium of the SRINM in plastic cages (OptiMice Biotech AS, 34 × 29 × 15 cm) in a temperature-controlled room (21–23 °C) with a reversed light-dark cycle (12h/12h; lights on 18:00 h; lights off 6:00 h) with food and water available ad libitum. This study was conducted following the recommendations of the European Communities Council Directive of September 22, 2010 (2010/63/EU). All experimental protocols were approved by the Ethics Committee of the SRINM.
2.2. Behavioural Tests
Behavioural tests were conducted between 9 am and 5 pm. Experimental mice have been acclimatized to the experimental room for 30 min. The behavioural equipment was cleaned with 70% ethanol between mice to remove residual odours. The subjective tests (open field, latent inhibition, and forced swim test) were video-recorded and analyzed by a skilled experimenter blind to genotype and drug treatment of the mice using EthoVision XT-10 software (Noldus Information Technology, Netherlands). Several cohorts of mice were used to minimize any influence of one behavioural procedure on another with minimum 7-days interval between the tests. The 1st cohort of mice was tested in the open field and forced swim test; 2nd – latent inhibition; and 3rd set of mice was used in PPI.
2.2.1. Open Field
Each mouse was placed in the middle of a bright lit (600 Lux) activity cage (Plexiglas cage; 40 cm × 40 cm × 37 cm) for 5 minutes. The total of travelled distance was assessed and analyzed.
2.2.2. Forced Swim Test (FST)
Each mouse was placed in a transparent plastic cylinder (25 cm high × 18 cm diameter) with water (25 °C) to the level of 18 cm and remained for 6 min. The time of immobility (floating, the state of navigation, when the mouse stopped moving and made only necessary movements to keep its head above water) was recorded during the last 4 minutes of the test period. After the experiment, each mouse was dried under a heated lamp and returned to the home cage afterwards.
2.2.3. Pre-Pulse Inhibition (PPI) of Acoustic Startle Response (ASR)
PPI was measured as previously described with mild modifications [13]. Six types of trials were used: 1) Pulse alone - single white noise burst (120dB 40ms); 2-5) Prepulse + pulse (PP72P, PP78P, PP82P, PP86P) –noise prepulse (20 ms at 69, 73, or 81dB) followed by startle pulse (120dB 40ms) 100ms after the prepulse onset; and 6) No-stimulus - background noise only (65dB). Sessions were structured as follows: 15-minute acclimation at background noise level; five Pulse trials; ten blocks each containing all six types of trials (Pulse, PP72P, PP78P, PP82P, PP86P, No-stimulus) in pseudorandom order; and five Pulse trials. The force intensity for each trial was recorded as the startling level. The percentage PPI induced by each prepulse intensity was calculated as [1-(startle amplitude on prepulse trial)/(startle amplitude on pulse alone)]*100%.
2.2.4. Latent Inhibition (LI)
The LI in the conditioning freezing paradigm was performed with mild modifications as previously described [35]. The experiment was conducted in a fear conditioning apparatus (Gemini avoidance system; San Diego Instruments). On the first day, half of the experimental animals, randomly assigned, received 40 presentations of a 30 sec pulsated tone (3.6 kHz, 80 dB) – the conditioned stimulus (CS) at a variable inter-stimulus interval of 40 ± 30 sec (pre-exposed group; PE). Immediately after 40th CS experimental mice received 5 pairings of CS-US (unconditioned stimulus; foot-shock, 0.4 mA; 1 sec), which was delivered at the end of CS with 3 minutes interval between CS-US pairings. Another half of mice received only 5 CS-US delivered after ~40 min spent in the operational chambers without exposure to PE (non-pre-exposed group; NPE). All mice were tested for the latent inhibition on the next day assessed in the open field coupled with a video-tracking system (Noldus Information Technology, Netherlands). Each mouse was placed in the centre of the open field (under regular light) and allowed to explore the novel environment for 3 minutes. Next, the CS (3.6 kHz, 80 dB) was delivered for the next 5 minutes without interruption from the audio-speakers located 1.5 m above the open field arena. Afterwards, CS was terminated and the mouse spent additional 2 minutes in the open field. Travelled distance during the whole testing session was analyzed for every 30 seconds.
2.3. Drugs
TC-2153 (8-(trifluoromethyl)benzo[f][1,2,3,4,5]pentathiepin-6-aminehydrochloride) was synthesized as described (Khomenko et al., 2011) with 99% purity. TC-2153 was dissolved in the distilled water with 2-3 drops of DMSO on the day of experiment [10 mg/kg] and injected 30 minutes before the behavioural test. In the LI experiment, TC-2153 was injected only on the 1st day, 30 minutes before the pre-exposition session to avoid any possible effect of the compound on the motor activity during the testing session.
2.4. Statistical Analysis
Statistical analysis was performed using the software package Statistica10 for Windows. Behavioral data were analyzed using one-way ANOVA, two-way ANOVA and two-way ANOVA with repeated measures (RM ANOVA), where necessary, with further post-hoc analysis (LSD Fisher test). Data are presented as Mean ± SEM.
3. Results
3.1. Effects of TC-2153 on Hyperactivity Assessed in the Open Field
Two-way ANOVA detected a main effect of genotype [F (1, 28) = 10.2; p < 0.001] and drug treatment [F (1, 28) = 19.3; p < 0.001] on travelled distance. Post-hoc analysis found that vehicle-treated Disc1-L100P mice walked significantly more than vehicle-treated WT littermates (p < 0.01). TC-2153 effectively reduced hyperactivity in Disc1-L100P mutant mice (p < 0.01) and also decreased ambulation in WT mice (p < 0.05) (Figure 2A).
3.2. Effects of TC-2153 on Behavioral Despair Assessed in the FST
Two-way ANOVA detected a main effect of genotype [F (1, 20) = 5.4; p < 0.05] and drug treatment [F (1, 20) = 6.4; p < 0.05] on the percentage of floating. Vehicle-treated Disc1-L100P floated significantly more than control WT mice (p < 0.05). TC-2153-treated mice of both genotypes significantly increased their activity (p’s < 0.05) in comparison with vehicle-treated mice (Figure 2B).
3.3. Effects of TC-2153 on Sensorimotor Gating Assessed by PPI of ASR
Two-way ANOVA with repeated measures found a main effect of pre-pulses [F (3, 63) = 40.3; p < 0.001] and genotype [F (1,21) = 16.1; p < 0.001] on startle response. Vehicle-treated Disc1-L100P mutants expressed PPI deficit at 74 dB (p < 0.01), 78dB, 82 dB and 86 dB (all p’s < 0.05) as compared to control WT mice. TC-2153 had no effect on PPI of mice of both genotypes (Figure 2C).
Two-way ANOVA detected a main effect of a genotype [F (1,21) = 21.4; p < 0.001] on the acoustic startle response. Post-hoc analysis found that vehicle-treated Disc1-L100P mice expressed lower ASR than control WT mice (p < 0.001). TC-2153 did not affect ASR in Disc1-L100P and WT mice (Table 1).
3.4. Effects of TC-2153 on Decremental Attention Assessed by LI
MANOVA with repeated measures detected a main effect of PE [F (1, 39) = 15.4; p < 0.001], genotype x drug interaction [F (1, 39) = 3.5; p < 0.05], time-interval [F (9, 351) = 2.4; p < 0.01], PE x time interval [F (9, 351) = 3.6; p < 0.05], time interval x genotype x PE interactions [F (9, 351) = 3.4; p < 0.05] on the percentage of freezing. Post-hoc analysis found that vehicle-treated PE-WT mice ignored the tone and travelled longer distance than NPE-WT group, demonstrating LI (p’s < 0.05 between 3rd and 4th minutes of CS’ exposure) (Figure 3A). In opposite, both PE and NPE groups of Disc1-L100P control mice behaved similarly, walking the comparable distance, expressing the disrupted LI (all p’s > 0.05) (Figure 3C). TC-2153-treated PE and NPE-WT mice showed no LI, walking similar distance (all p’s > 0.05) (Figure 3B). In contrast to WT, STEP inhibitor given to Disc1-L100P ameliorated their LI deficit, increasing travelled distance in PE group in comparison with vehicle PE-Disc1-L100P mutants and TC-2153-treated NPE-Disc1-L100P mice (Figure 3D).
4. Discussion
Our pre-clinical findings support STEP as a promising molecular target for the development of next-generation antipsychotic drugs (APDs), capable of eliciting therapeutic effects in a gene-dependent manner and advancing the field of personalized psychiatry. Notably, TC-2153 affected motor activity similarly in both WT and Disc1-L100P mutant mice, as assessed in the open field and FST. However, it selectively corrected LI deficit in Disc1-L100P mutants while impairing LI in WT mice. This suggests that the APD-like effects of STEP inhibition are DISC1-dependent, as evident by its selective action on LI. Importantly, TC-2153 also induced side effects in WT mice, including reduced ambulation in the open field, increased activity under the stressful conditions in the FST, and disrupted LI. These observations reinforce the gene-dependent effectiveness of STEP inhibition and highlight its potential contribution to personalized medicine approaches in psychiatry.
The etiology of schizophrenia is complex, and understanding its molecular mechanisms is a critical step toward effective drug discovery. Rapid advances in functional genomics – particularly in the regulation of transcription and spatial configuration of DNA – have opened new avenues for identifying novel drug targets and even synergistic therapies that can simultaneously modulate multiples molecular pathways [36,37]. Despite these advances, we are still in the early stages of determining how gene discovery will inform personalized treatment strategies for schizophrenia. Considerable efforts have been made to uncover the genetic factors contributing to variability in APD responsiveness and the genetic risk of developing side effects, with the ultimate goal of improving treatment effectiveness [38].
The generations of genetically engineered mouse models have provided researchers with a powerful toolbox for pharmacogenetic studies of schizophrenia. Among these, Disc1-L100P mutant mice meet all three criteria for a valid animal model of schizophrenia: construct, face and predictive validity [13]. Construct validity is supported by multiple independent genetic studies linking polymorphisms in the DiISC1 gene to schizophrenia [39]. Notably, the W160L mutation detected in patients with schizophrenia is closely related to the L100P mutation used in this mouse model [40]. Furthermore, we showed a pathogenic interaction between Disc1-L100P and maternal immune activation [41], aligning with the gene-environment interplay implicated in schizophrenia pathogenesis [42]. Face validity is reflected in the schizophrenia-like behavioral phenotypes in Disc1-L100P mice, including hyperactivity, deficient PPI and LI, found in the current study and supporting previous reports [11,12,13,35,43,44]. Our current study also revealed increased floating in Disc1-L100P mutant mice assessed in FST, suggesting reduced motivation to cope with aversive conditions, which may correspond to negative symptoms of schizophrenia. The emergence of this new phenotype in Disc1-L100P mutant mice warrants further investigation and may be influences by differences in genetic background between the original and current Disc1-L100P lines, including the number of backcrosses to the C57BL/6J strain, experimental conditions, or housing environments. Predictive validity is demonstrated by the response of Disc1-L100P mice to antipsychotic treatment. As previously reported, clozapine haloperidol - but not bupropion - were able to correct hyperactivity and PPI deficit without affecting startle response and restored LI deficit without impacting long-term memory [11,13]. Notably, clozapine was more effective than haloperidol in Disc1-L100P mice [43], supporting the dopaminergic hypersensitivity driven by altered Disc1 - D2R - GSK-3 protein x protein interactions [11,12].
We investigated the effectiveness of the STEP inhibitor TC-2153 in the Disc1-L100P mouse model of schizophrenia for the first time. TC-2153 reduced hyperactivity in Disc1 mutant mice to levels comparable to TC-2153-treated WT mice, aligning well with the effects observed for other compounds such as the PDE4 inhibitor rolipram [13], the GSK-3 inhibitor TDZD-8 [12], and the Disc1-D2R uncoupling peptide [11]. In addition, to its impact on locomotion in the open field, TC-2153 also reduced floating behavior in mice of both genotypes, supporting its potential antidepressant effect [28]. However, further studies are needed to distinguish between negative symptoms of schizophrenia-related behaviour and depression-like phenotypes in Disc1-L100P mice.
Interestingly, TC-2153 did not alter the PPI deficit in Disc1-L100P mice, however, the STEP inhibitor effectively ameliorated their LI deficit. This distinct effect stems from the fact that PPI and LI assess different cognitive processes and are regulated by specific underlying neural pathways and biochemical mechanisms. PPI measures sensorimotor gating, referring to the attenuation of the startle response when a weak pre-pulse precedes the startle stimulus by a short interval. LI reflects the ability to learn to ignore irrelevant stimuli (conditioned stimulus; CS) and has been extensively studied in both clinical and animal research [45]. Briefly, animals pre-exposed (PE) to the CS without reinforcement learnt to disregard the CS-unconditioned stimulus (US) association. LI is quantified as the difference in learning between PE and non-pre-exposed (NPE) groups.
Our current study found disrupted LI in Disc1-L100P mice, consistent with recent findings using a fear conditioning paradigm [35]. Both PPI and LI deficits are well-established phenotypes in schizophrenia patients [46,47,48,49]. Notably, dopaminergic (DA) agonists and NMDA antagonists can disrupt PPI [50], and LI [51], albeit through different mechanisms. For example, amphetamine, a DA releaser, disrupts both PPI and LI in rodents, while NMDA receptor antagonists disrupt PPI but typically spare LI. Amphetamine-induced deficits in PPI and LI can be reversed by both typical and atypical APDs [46,51,52], with atypical APDs showing greater effectiveness against NMDA antagonists-induced PPI and LI abnormalities.
Given that LI disruption in Disc1-L100P mice was rescued by DA-related compounds (haloperidol, PDE4B- and GSK-3- inhibitors, DISC1 x D2R uncoupling peptide) as we previously demonstrated - we propose that TC-2153 may exert its effects via DA-related mechanisms. This may also explain the disruptive effect of the STEP inhibitor on LI of WT mice, resembling the action of amphetamine. Therefore, the lack of effectiveness of TC-1253 on the PPI deficit in Disc1-L100P mice might be linked to glutamatergic pathways specifically involved in sensorimotor gating.
Indeed, accumulating pharmacological [26] and genetic [53] evidence supports NMDA-dependent mechanisms in STEP signaling. For example, TC-2153 was shown to reverse hyperactivity induced by the NMDAR antagonist PCP in mice [26], while genetic knockdown of the STEP gene increased sensitivity to the NMDAR blocker MK-801 [53]. STEP functions by dephosphorylating and thereby inactivating key signaling proteins involved in synaptic strengthening, including the NR2B subunit of the NMDAR complex [54].
Although the molecular dynamics of the NMDAR complex and associated synaptic plasticity in excitatory neurons have not yet been studied in our Disc1 mutant mice, recent studies suggest that DISC1 regulates NMDAR activity through its interaction with the NR1 subunit [55]. Additionally, reduced excitatory synaptic transmission has been observed in the hippocampus of Disc1-L100P mice [56], indicating potential hypofunction in NMDAR-mediated neurotransmission.
These findings underscore the urgent need to further explore the interplay between DISC1 and STEP in regulating glutamatergic and dopaminergic neurotransmission, especially withing specific neural subpopulations involved in modulating PPI and LI processes.
5. Conclusions
Overall, the STEP inhibitor TC-2153 effectively reversed schizophrenia-like behavioral phenotypes in Disc1-L100P mutant mice, including hyperactivity and disrupted LI. In contrast, TC-2153 impaired LI and reduced motor activity in WT mice. Notably, the STEP inhibitor also induced antidepressant-like action regardless of genotype. These findings support STEP as a promising target for APD development and suggest a potential link between STEP and the DISC1 molecular complex in the pathogenesis of schizophrenia – a connection that warrants further investigation. Finally, our data indicates that the development of new STEP inhibitors could advance pharmacogenetic strategies, paving the way for personalized psychiatric therapy.
Author Contributions
T.V.L., A.A.T., T.G.A. designed experiments; A.A.T. performed behavioral experiments; T.M.K., K.P.V. synthesized TC-2153; T.V.L. analyzed the data; T.V.L., T.G.A., K.P.V., N.F.S. interpreted the data; T.L. and A.A.T. wrote the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the Siberian Branch of Russian Academy of Science program for the Fundamental Interdisciplinary Integrative Research (2018-2020); “Unique scientific installation “Biological collection – Genetic biomodels of neuro-psychiatric disorders” (№ 493387) Federal State Budgetary Scientific Institution «Scientific Research Institute of Neurosciences and Medicine». All authors report no financial interests or potential conflicts of interest.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The original contributions presented in this study are included in the article. Further inquiries can be directed at the corresponding author.
Acknowledgments
We are thankful to Konstantin Pavlov for his skillful technical assistance with EthoVision software. We thank Lubov Ruban and Svetlana Tataurova for the preparation of experimental mice and animal husbandry.
Conflicts of Interest
The authors declare no competing interests.
References
- Snyder, S.H. The dopamine hypothesis of schizophrenia: focus on the dopamine receptor. Am J Psychiatry 1976, 133, 197-202. [CrossRef]
- Howes, O.D., Kapur, S. The dopamine hypothesis of schizophrenia: version III – the final common pathway. Schizophr Bull, 2009, 35, 549-562. [CrossRef]
- Howes, S.R., Dalley, J.W., Morrison, C.H., Robbins, T.W., Everitt, B.J. Left ward shift in the acquisition of cocaine self-administration in isolation– reared rats: relationship to extracellular levels of dopamine, serotonin and glutamate in the nucleus accumbens and amygdala– striatal FOS expression. Psychopharmacology (Berl) 2000, 151, 55-63. [CrossRef]
- Jones, G.H., Hernandez, T.D., Kendall, D.A., Marsden, C.A., Robbins, T.W. Dopaminergic and serotonergic function following isolation rearing in rats: study of behavioural responses and postmortem and in vivo neurochemistry. Pharmacol Biochem Behav 1992, 43, 17-35. [CrossRef]
- Pruessner, J.C., Champagne, F., Meaney, M.J., Dagher, A. Dopamine release in response to a psychological stress in humans and its relationship to early life maternal care: a positron emission tomography study using [11C]raclopride. J Neurosci 2004, 24, 2825-2831. [CrossRef]
- GBD 2017. Disease and injury incidence and prevalence collaborators. Global, regional, and national incidence, prevalence, and years lived with disability for 354 diseases and injuries for 195 countries and territories, 1990-2017: a systematic analysis for the Global Burden of Disease Study 2017. Lancet, 2018, 392, 1789-1858. [CrossRef]
- Seeman, P., Lee, T., Chau-Wong, M., Wong, K. Antipsychotic drug does and neuroleptic/dopamine receptors. Nature 1976, 261, 717-719. [CrossRef]
- Missale, C., Nash, S.R., Robinson, S.W., Robinson, S.W., Jaber, M., Caron, M.G. Dopamine receptors: from structure to function. Physiol Rev 1998, 78, 189-225. [CrossRef]
- Beaulieu, J-M, Gainetdinov, R.R., Caron, M.G. Akt/GSK3 signaling in the action of psychotropic drugs. Annual Rev Pharmacol Toxicol 2009, 49, 327-347. [CrossRef]
- Hanyaloglu, A.C., von Zatrow, M. Regulation of GPCRs by endocytic membrane trafficking and its potential implications. Annual Rev Pharmacol Toxicol 2008, 48, 537 – 568. [CrossRef]
- Su, P., Li, S., Chen, S., Lipina, T.V., Wang, M., Lai, T.K., Lee, F.H., Zhang, H., Zhai, D., Ferguson, S.S., Nobrega, J.N., Wong, A.H., Roder, J.C., Fletcher, P.J., Liu, F. A dopamine D2 receptor-DISC1 protein complex may contribute to antipsychotic-like effects. Neuron 2014, 4, 1302-1316. [CrossRef]
- Lipina, T.V., Kaidanovich-Beilin, O., Patel, S., Wang, M., Clapcote, S.J., Liu, F., Woodgett, J.R., Roder, J.C. Genetic and pharmacological evidence for schizophrenia-related Disc1 interaction with GSK-3. Synapse 2011, 65, 234-248. [CrossRef]
- Clapcote, S.J., Lipina. T.V., Millar, J.K., Mackie, S., Christie, S., Ogawa, F., Lerch, J.P., Trimble, K., Uchiyama, M., Sakuraba, Y., Kaneda, H., Shiroishi, T., Houslay, M.D., Henkelman, R.M., Sled, J.G., Gondo, Y., Porteous, D.J., Roder, J.C. Behavioral phenotypes of Disc1 missense mutations in mice. Neuron 2007, 54, 387-402. [CrossRef]
- Dahoun, T., Trossbach, S.V., Brandon, N.J., Korth, C., Howes, O.D. The impact of Disrupted-In-Schizophrenia (DISC1) on the dopaminergic system: a systematic review. Transl Psychiatry 2017, 7, e1015. [CrossRef]
- Carty, N.C., Xu, J., Kurup, P., Brouillette, J., Goebel-Goody, S.M., Austin, D.R., Yuan, P., Chen, G., Correa, P.R., Haroutunian, V., Pittenger, C., Lombroso, P.J. The tyrosine phosphatase STEP: implications in schizophrenia and the molecular mechanism underlying antipsychotic medications. Transl Psychiatry 2012, 2, e137. [CrossRef]
- Lombroso, P.J., Murdoch, G., Lerner, M. Molecular characterization of a protein-tyrosine-phosphatase enriched in striatum Proc Natl Acad Sci USA 1991, 88, 7242-7246. [CrossRef]
- Kim, S.Y., Lee, H.J., Kim, Y.N., Yoon, S., Lee, J.E., Sun, W., Choi, E.J., Baik, J.H. Striatal-enriched protein tyrosine phosphatase regulates dopaminergic neuronal development via extracellular signal-regulated kinase signaling. Exp Neurol 2008, 214, 69-77. [CrossRef]
- Snyder, E.M., Nong, Y., Almeida, C.G., Paul, S., Moran, T., Choi, E.Y., Nairn, A.C., Salter, M.W., Lombroso, P.J., Gouras, G.K., Greengard, P. Regulation of NMDA receptor trafficking by amyloid-beta. Nat Neurosci 2005, 8, 1051-1058. [CrossRef]
- Zhang, Y., Venkitaramani, D.V., Gladding, C.M., Zhang, Y., Kurup, P., Molnar, E., Collingridge, G.L., Lombroso, P.J. The tyrosinephosphatase STEP mediates AMPA receptor endocytosis after metabotropic glutamate receptor stimulation. J Neurosci 2008, 28, 10561–10566. [CrossRef]
- Paul, S., Nairn, A.C., Wang, P., Lombroso, P.J. NMDA-mediated activation of the tyrosine phosphatase STEP regulates the duration of ERK signaling. Nat Neurosci 2003, 6, 34-42. [CrossRef]
- Nguyen, T.H., Liu, J., Lombroso, P.J. Striatal enriched phosphatise 61 dephosphorylates Fyn at phosphotyrosine 420. J Biol Chem 2002, 277, 24274–24279. [CrossRef]
- Xu, J., Kurup, P., Bartos, J.A., Patriarchi, T., Hell, J.W., Lombroso, P.J. STriatal-enriched protein tyrosine phosphatase (STEP) regulates Pyk2 activity. J Biol Chem 2012, 287, 20942-20956. [CrossRef]
- Goebel-Goody, S.M., Baum, M., Paspalas, C.D., Fernandez, S.M., Carty, N.C., Kurup, P., Lombroso, P.J. Therapeutic implications forstriatal-enriched protein tyrosine phosphatase (STEP) in neuropsychiatric disorders. Pharmacol Rev, 2012, 64, 65-87. [CrossRef]
- Khomenko, T.M., Tolstikova, T.G., Bolkunov, A.V., Dolgikh, M.P., Pavlova, A.V., Korchagina, D.V., Volcho, K.P., Salakhutdinov, N.F. 8–(Trifluoromethyl)–1,2,3,4,5–benzopentathiepin–6–amine: novel aminobenzopentathiepine having in vivo anticonvulsant and anxiolytic activities. Lett Drug Design Discov 2009, 6, 464-467. [CrossRef]
- Baguley, T.D.; Nairn, A.C., Lombroso, P.J., Ellman, J.A. Synthesis of benzopentathiepin analogs and their evaluation as inhibitor of the phosphatase STEP. Bioorg Med Chem Lett 2015, 25,1044-1046. [CrossRef]
- Xu, J., Kurup, P., Baguley, T.D., Foscue, E., Ellman, J.A., Nairn, A.C., Lombroso, P.J. Inhibition of the tyrosine phosphate STEP61 restores BDNF expression and reverses motor and cognitive deficits in phencyclidine-treated mice. Cell Mol Life Sci 2016, 73, 1503-1514. [CrossRef]
- Zong, M-m., Yuan, H-m., He, X., Zhou, Z-q., Qiu, X-d., Yang, J-j., Ji, M-h. Disruption of Striatal-Enriched protein tyrosine phosphatise signalling might contribute to memory impairment in a mouse model of sepsis-associated encephalopathy. Neurochem Res 2019, 44, 2832-2842. [CrossRef]
- Kulikova, E.A., Volcho, K.P., Salakhutdinov, N.F., Kulikov, A.V. Benzopentathiepine derivative, 8-(trifluoromethyl)-1,2,3,4,5-benzopentathiepin-6-amine hydrochloride (TC-2153), as a promising antidepressant of new generation. Lett Drug Design Discov 2017, 14, 974-984. [CrossRef]
- Chatterjee, M., Singh. P., Xu. J., Lombroso. P.J., Kurup. P.K. Inhibition of striatal-enriched protein tyrosine phosphatise (STEP) activity reverses behavioural deficits in a rodent model of autism. Behav Brain Res 2020, 391, 112713. [CrossRef]
- Moskaliuk, V.S., Kozhemyakina, R.V., Bazovkina, D.V., Terenina, E., Khomenko, T.M., Volcho, K.P., Salakhutdinov, N.F., Kulikov, A.V., Naumenko, V.S., Kulikova, E. Biomedicine & Pharmacotherapy 2022, 147, 112667. [CrossRef]
- Moskaliuk, V.S., Kozhemyakina, R.V., Khomenko, T.M., Volcho, K.P., Salakhutdinov, N.F., Kulikov, A.V., Naumenko, V.S., Kulikova, E. On Associations between Fear-Induced Aggression, Bdnf Transcripts, and Serotonin Receptors in the Brains of Norway Rats: An Influence of Antiaggressive Drug TC-2153. Int. J. Mol. Sci, 2023, 24, 983. [CrossRef]
- Moskaliuk, V.S., Kozhemyakina, R.V., Khomenko, T.M., Volcho, K.P., Salakhutdinov, N.F., Kulikov, A.V., Naumenko, V.S., Kulikova, E. Key Enzymes of the Serotonergic System - Tryptophan Hydroxylase 2 and Monoamine Oxidase A – In the Brain of Rats Selectively Bred for a Reaction toward Humans: Effects of Benzopentathiepin TC-2153. Biochemistry (Moscow),2024, 89, 1109-1121. [CrossRef]
- Lipina, T.V., Roder, J.C. Disrupted-In-Schizophrenia-1 (DISC1) interactome and mental disorders: impact of mouse models. Neurosci Biobehav Rev 2014, 45, 271-294. [CrossRef]
- Petrova, E.S., Gromova, A.V., Anisimenko, M.S., Ruban, L.A., Egorova, S.A., Petrovskaya, E.F., Amstislavskaya, T.G., Lipina, T.V. Maintenance of genetically modified mouse lines: input to the development of bio-collections in Russia. Laboratory animals for science, 2018, 2, 2-15. [CrossRef]
- Lipina, T.V., Beregovoy, N.A., Tkachenko, A.A., Petrova, E.S., Starostina, M.V., Zhou, Q., Li, S. Uncoupling DISC1 x D2R protein-protein interactions facilitates latent inhibition in Disc1-L100P animal models of schizophrenia and enhances synaptic plasticity via D2 receptors. Frontiers Synaptic Neuroscience 2018, 10, 31. [CrossRef]
- Rajarajan, P., Gil, S.E., Brennand, K.J., Akbarian, S. Spatial genome organization and cognition. Nat Rev Neurosci 2016, 17, 681-691. [CrossRef]
- Han, K., Jeng, E.E., Hess, G.T., Morgens, D.W., Li, A., Bassik, M.C. Synergistic drug combinations for cancer identified in a CRISPR screen for pairwise genetic interactions. Nat Biotechnol 2017, 35, 463-474. [CrossRef]
- Foley, C., Corvin, A., Nakagome, S. Genetics of schizophrenia: ready to translate? Curr Psychiatry Rep 2017, 19, 61. [CrossRef]
- Bradshaw, N.J., Porteous, D.J. DISC1-binding proteins in neural development, signaling and schizophrenia. Neuropharmacology 2012, 62, 1230-1241. [CrossRef]
- Thomson, P.A., Parla, J.S., McRae, A.F., Kramer, M., Ramakrishnan, K., Yao, J., Soares, D.C., McCarthy, S., Morris, S.W., Cardone, L., Cass, S., Ghiban, E., Hennah, W., Evans, K.L., Rebolini, D., Millar, J.K., Harris, S.E., Starr, J.M., MacIntyre, D.J., Generation Scotland, McIntosh, A.M., Watson, J.D., Deary, I.J., Visscher, P.M., Blackwood, D.H., McCombie, W.R., Porteous, D.J. 708 Common and 2010 rare DISC1 locus variants identified in 1542 subjects: analysis for association with psychiatric disorder and cognitive traits. Mol Psychiatry 2014, 19, 668-675. [CrossRef]
- Lipina, T.V., Zai, C., Hlousek, D., Roder, J.C., Wong, A.H. Maternal immune activation during gestation interacts with Disc1 point mutation to exacerbate schizophrenia-related behaviors in mice. J Neurosci 2013, 33, 7654-7666. [CrossRef]
- Lewis, D.A., Levitt, P. Schizophrenia as a disorder of neurodevelopment Annu Rev Neurosci 2002, 25, 409-432. [CrossRef]
- Lipina, T.V., Niwa, M., Jaaro-Peled, H., Fletcher, P.J., Seeman, P., Sawa, A,. Roder, J.C. Enhanced dopamine function in DISC1-L100P mutant mice: implications for schizophrenia. Genes Brain Behav 2010, 9, 777-789.
- Walsh, J., Desbonnet, L., Clarke, N., Waddington, J.L., O’Tuathaigh, C.M. Disruption of exploratory and habituation behavior in mice with mutation of DISC1: an ethologically based analysis. J Neurosci Res 2012, 90, 1445-1453. [CrossRef]
- Lubow, R.E. A short history of latent inhibition research. In the book “Latent inhibition: cognition, neuroscience and applications to schizophrenia”. 2010, p. 1-10; Eds R.E. Lubow and I. Weiner. Cambridge University Press. [CrossRef]
- Swerdlow, N.R., Braff, D.L., Taaid, N., Geyer, M.A. Assessing the validity of an animal model of deficient sensorimotor gating in schizophrenia patients. Arch Gen Psychiatry 1994, 51, 139-154. [CrossRef]
- Braff, D., Geyer, M.A., Swerdlow, N.R. Sensorimotor gating and schizophrenia: human and animal model studies. Arch Gen Psychiatry 2001, 47, 181–188. [CrossRef]
- Weiner, I., Shadach, E., Tarrasch, R., Kidron, R., Feldon, J. The latent inhibition model of schizophrenia: further validation using the atypical neuroleptic, clozapine. Biol Psychiatry 1996, 40, 834-843. [CrossRef]
- Gray, N.S., Hemsley, D.R., Gray, J.A. Abolition of latent inhibition in acute, but not chronic, schizophrenics. Neurol PsychiatryBrain Res, 1992, 1, 83-89. [CrossRef]
- Geyer, M.A., Krebs-Thomson, K., Braff, D.L., Swerdlow, N.R. Pharmacological studies of prepulse inhibition models sensorimotor gating deficits in schizophrenia: a decade in review. Psychopharmacology, 2001, 156, 117-154. [CrossRef]
- Moser, P.C., Hitchcock, J.M., Lister, S., Moran, P.M. The pharmacology of latent inhibition as an animal model of schizophrenia. Brain Res Rev 2000, 33, 275-307. [CrossRef]
- Weiner, I., Feldon, J. The switching model of latent inhibition: an update of neural substrate. Beh Brain Res 1997, 88, 11-25. [CrossRef]
- Sukoff Rizzo, S.J., Lotarski, S.M., Stolyar, P., McNally, T., Arturi, C., Roos, M., Finely, J.E., Reinhart, V., Lanz, T.A. Behavioral characterization of striatal-enriched protein tyrosine phosphatase (STEP) knockout mice. Genes Brain Behav 2014, 13, 643-652. [CrossRef]
- Fitzpatrick, C.J., Lombroso, P.J. The role of striatal-enriched protein tyrosine phosphatase (STEP) in cognition. Front Neuroanatomy 2011, 5, 47. [CrossRef]
- 55. Malavasi, E.L.V., Economides, K.D., Grunewald, E., Makedonopoulou, P., Gautier, P., Mackie, S., Murphy, L.C., Murdoch, H., Crummie, D., Ogawa, F., McCartney, D.L., O’Sullivan, S.T., Burr, K., Torrance, H.S., Phillips, J., Bonneau, M., Anderson S.M., Perry, P., Pearson, M., Costantinidies, C., Davidson-Smith, H., Kabiri, M., Duff, B., Johnstone, M., Polites, H.G., Lawrie, S.M., Blackwood, D.H., Semple, C.A., Evans, K.L., Didier, M., Chandran, S., McIntosh, A.M., Price, D.J., Houslay, M.D., Porteous, D.J., Millar, J.K. DISC1 regulates N-methyl-D-asparate receptor dynamics: abnormalities induced by a Disc1 mutation modelling a translocation linked to major mental illness. Transl Psychiatry 2018, 8, 184. [CrossRef]
- Cui, L., Sun, W., Yu, M., Li, N., Guo, L., Gu, H., Zhou, Y. Disrupted-in-schizophrenia1 (DISC1) L100P mutation alters synaptic transmission and plasticity in the hippocampus and causes recognition memory deficits. Mol Brain 2016, 9, 89. [CrossRef]
Figure 1.
Chemical structure of the STEP inhibitor, TC-2153, (8-(trifluoromethyl)benzo[f][1,2,3,4,5]pentathiepin-6-amine hydrochloride).
Figure 1.
Chemical structure of the STEP inhibitor, TC-2153, (8-(trifluoromethyl)benzo[f][1,2,3,4,5]pentathiepin-6-amine hydrochloride).
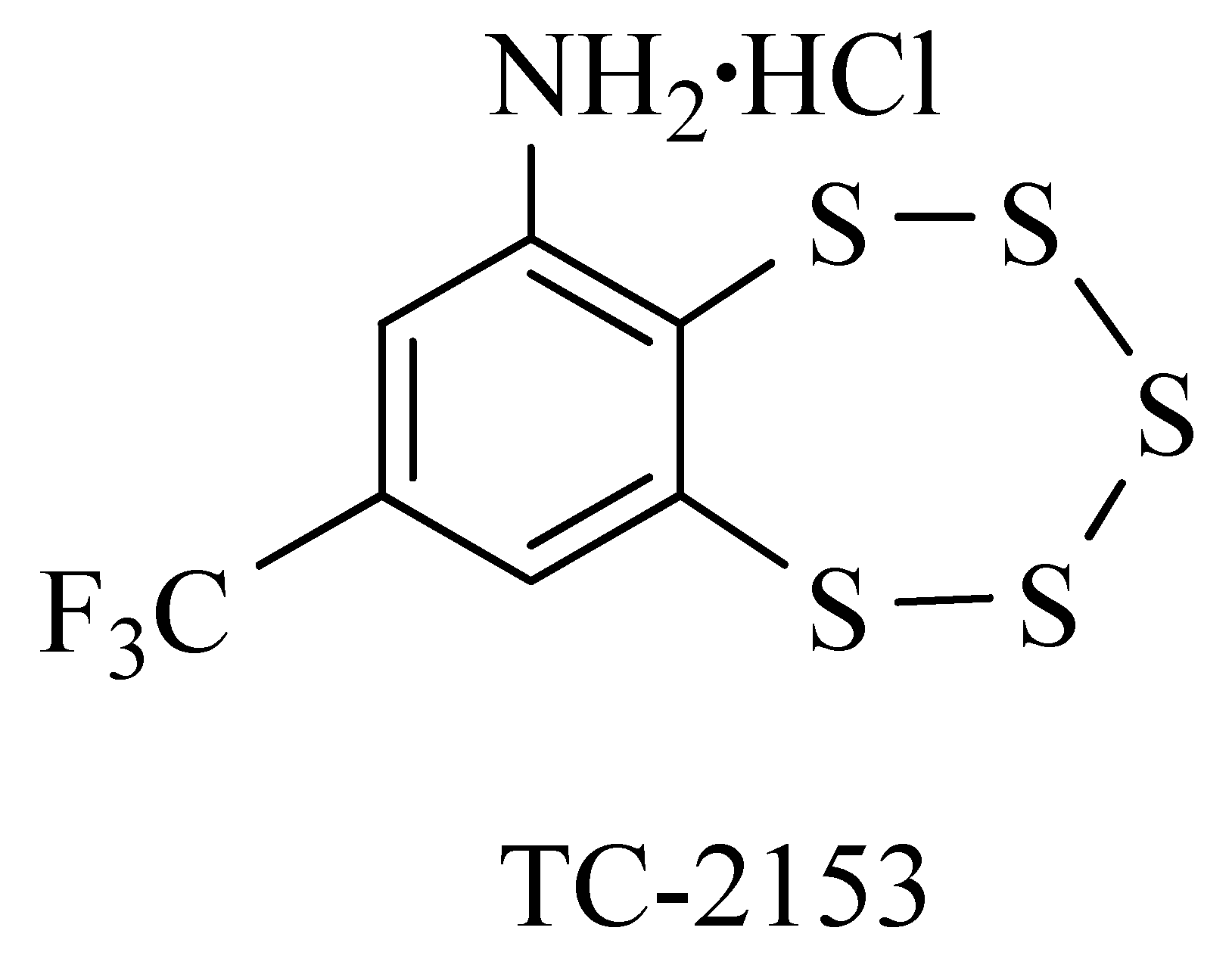
Figure 2.
A-C. TC-2153 reduced motor activity assessed in the open field (A); reduced duration of floating (%) in the Forced Swim Test (B) in both wild-type (WT) and Disc1-L100P mutant mice and had no effect on the percentage of pre-pulse inhibition of acoustic startle response (PPI) (C). * - p < 0.05; ** - p < 0.01 – in comparison vehicle-treated mice within each experimental group; # - p < 0.05; ## - p < 0.01 – in comparison with vehicle-treated WT mice. N = 6-8 mice per group.
Figure 2.
A-C. TC-2153 reduced motor activity assessed in the open field (A); reduced duration of floating (%) in the Forced Swim Test (B) in both wild-type (WT) and Disc1-L100P mutant mice and had no effect on the percentage of pre-pulse inhibition of acoustic startle response (PPI) (C). * - p < 0.05; ** - p < 0.01 – in comparison vehicle-treated mice within each experimental group; # - p < 0.05; ## - p < 0.01 – in comparison with vehicle-treated WT mice. N = 6-8 mice per group.
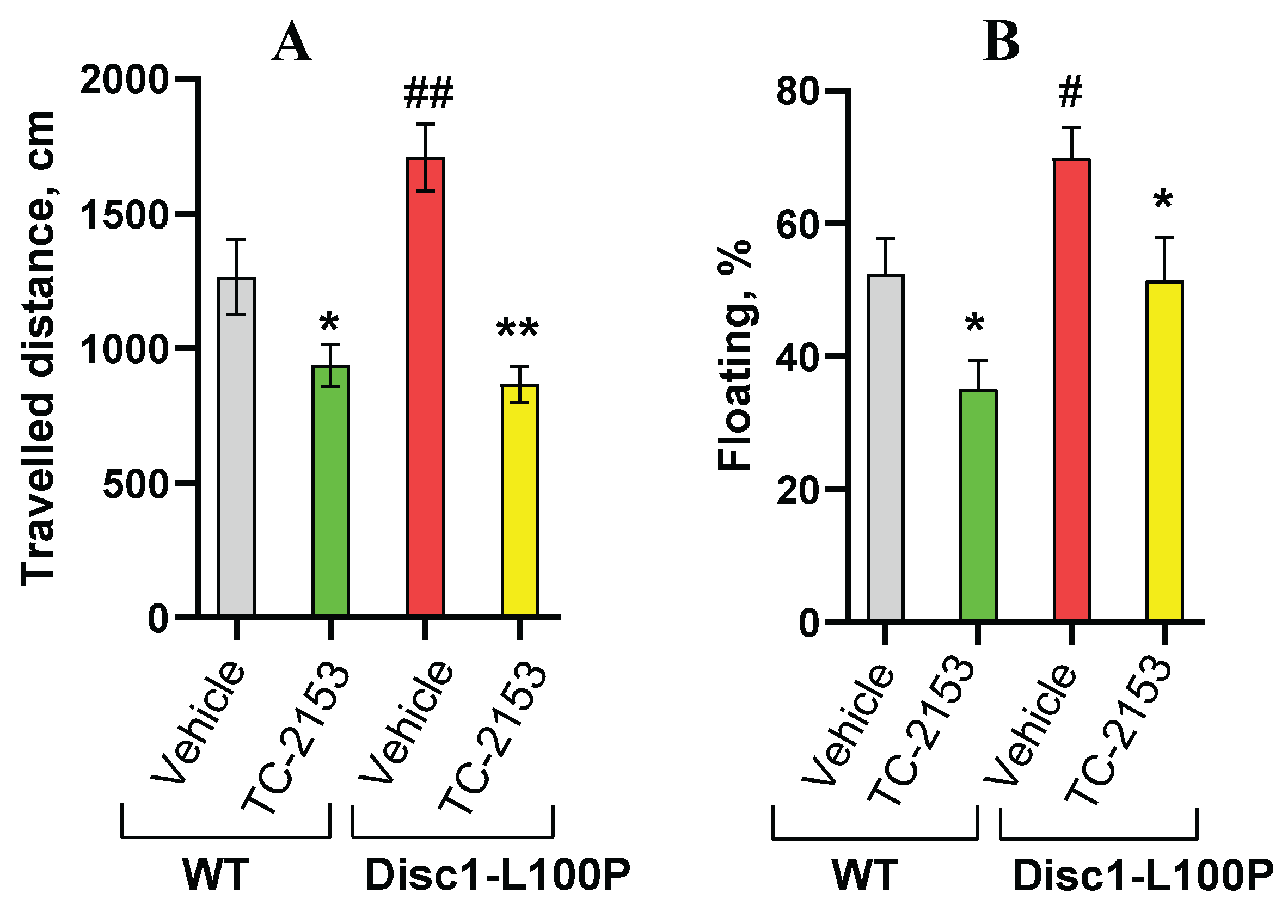
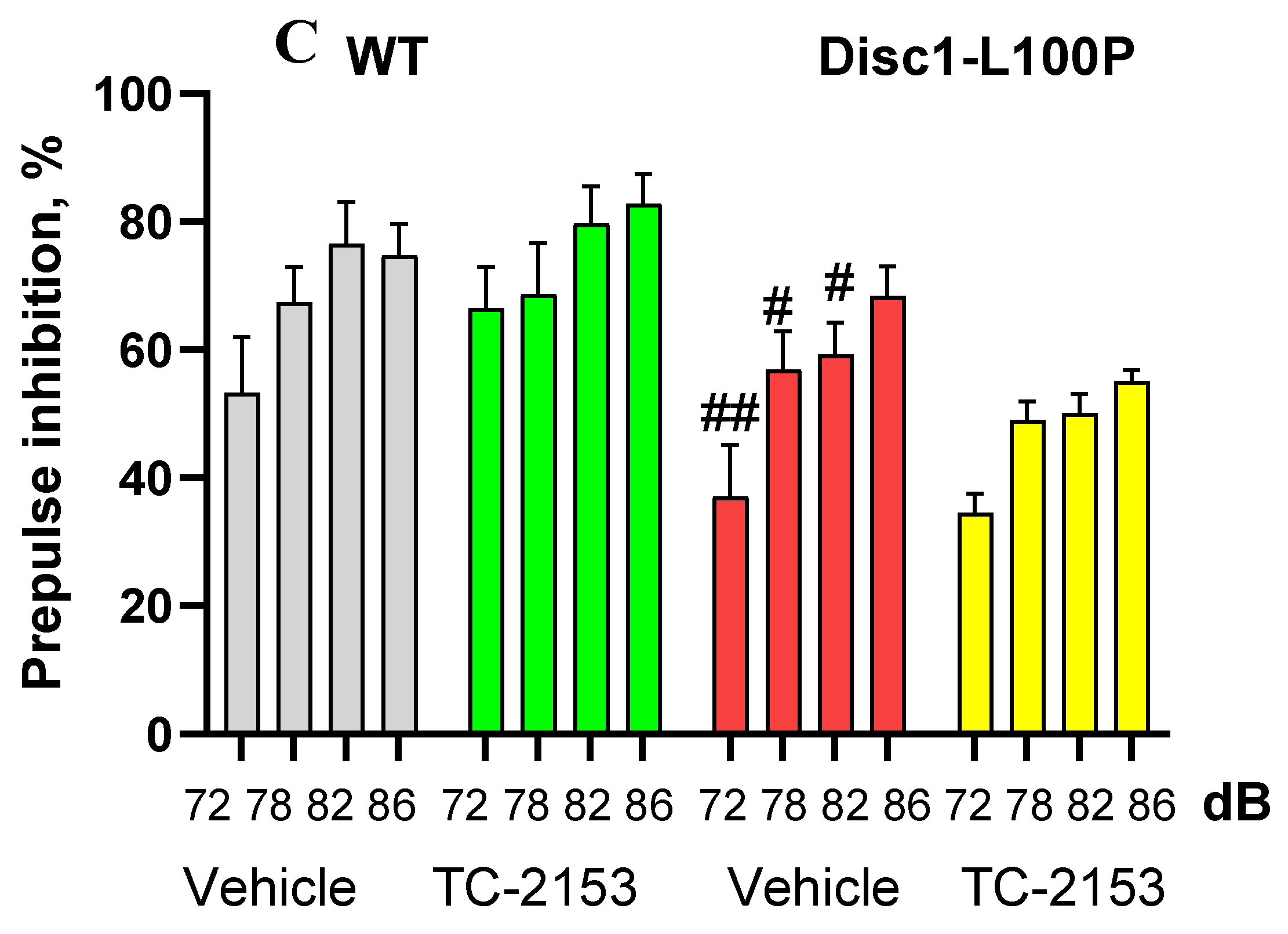
Figure 3.
A-D. TC-2153 corrected disrupted latent inhibition (LI) in Disc1-L100P mice. (A) vehicle-treated wild-type (WT) mice pre-exposed (PE) to the conditioned stimulus (CS) did not decrease their ambulation in response to CS (grey colored line) in opposite to the non-preexposed (NPE) group of control mice, whereby showing LI, which was disrupted by TC-2153 (B). Vehicle-treated Disc1-L100P mutant mice did not express LI (C), however, TC-2153 significantly improved the expression of LI in Disc1-L100P mice (D) by increasing travelled distance in PE-Disc1-L100P mutants. * - p < 0.05; ** - p < 0.01 – in comparison with NPE group within each experimental group; # - p < 0.05; ## - p < 0.01; ### - p < 0.001 – in comparison with vehicle-treated Disc1-L100P mice. N = 7-8 mice per group.
Figure 3.
A-D. TC-2153 corrected disrupted latent inhibition (LI) in Disc1-L100P mice. (A) vehicle-treated wild-type (WT) mice pre-exposed (PE) to the conditioned stimulus (CS) did not decrease their ambulation in response to CS (grey colored line) in opposite to the non-preexposed (NPE) group of control mice, whereby showing LI, which was disrupted by TC-2153 (B). Vehicle-treated Disc1-L100P mutant mice did not express LI (C), however, TC-2153 significantly improved the expression of LI in Disc1-L100P mice (D) by increasing travelled distance in PE-Disc1-L100P mutants. * - p < 0.05; ** - p < 0.01 – in comparison with NPE group within each experimental group; # - p < 0.05; ## - p < 0.01; ### - p < 0.001 – in comparison with vehicle-treated Disc1-L100P mice. N = 7-8 mice per group.
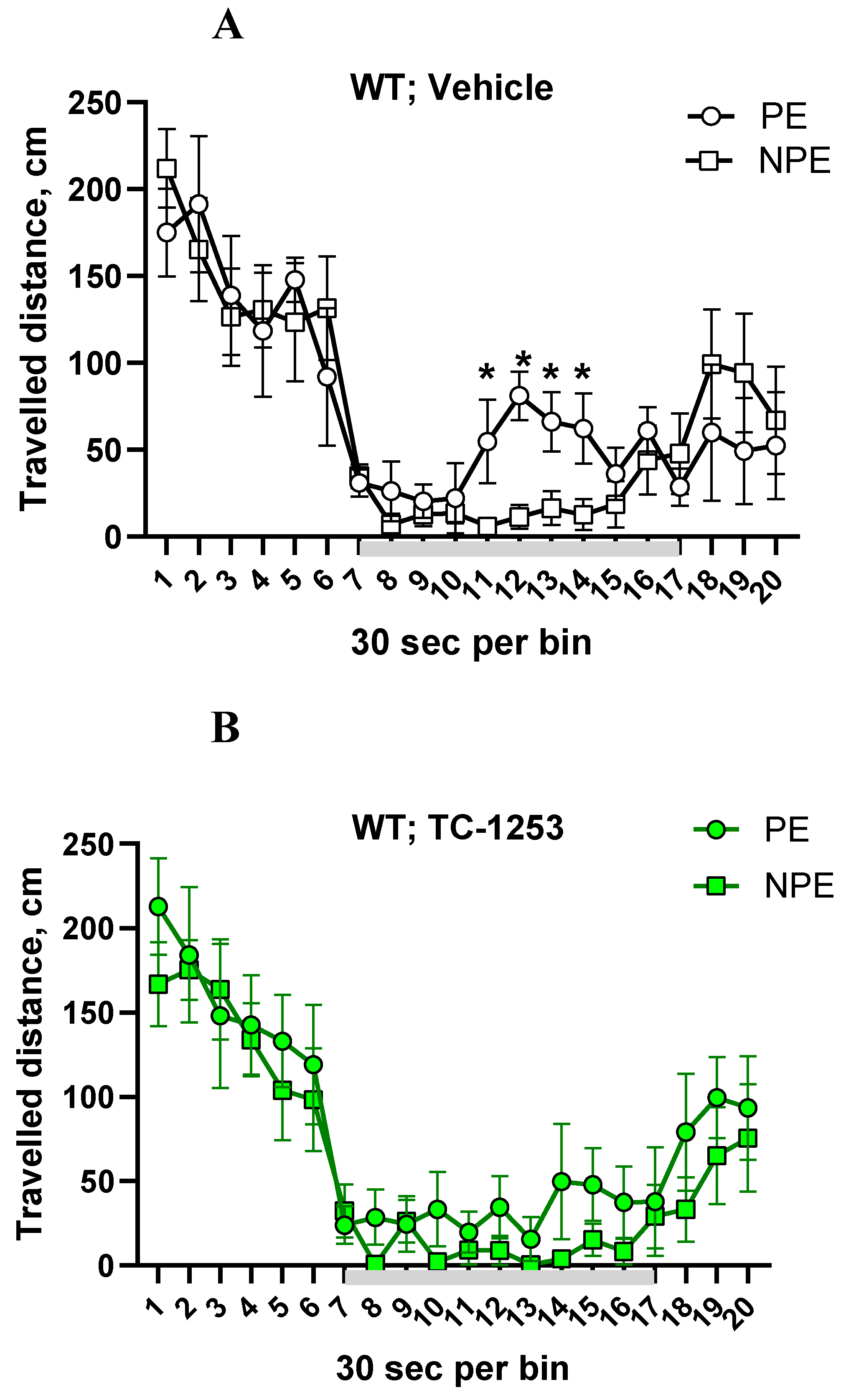
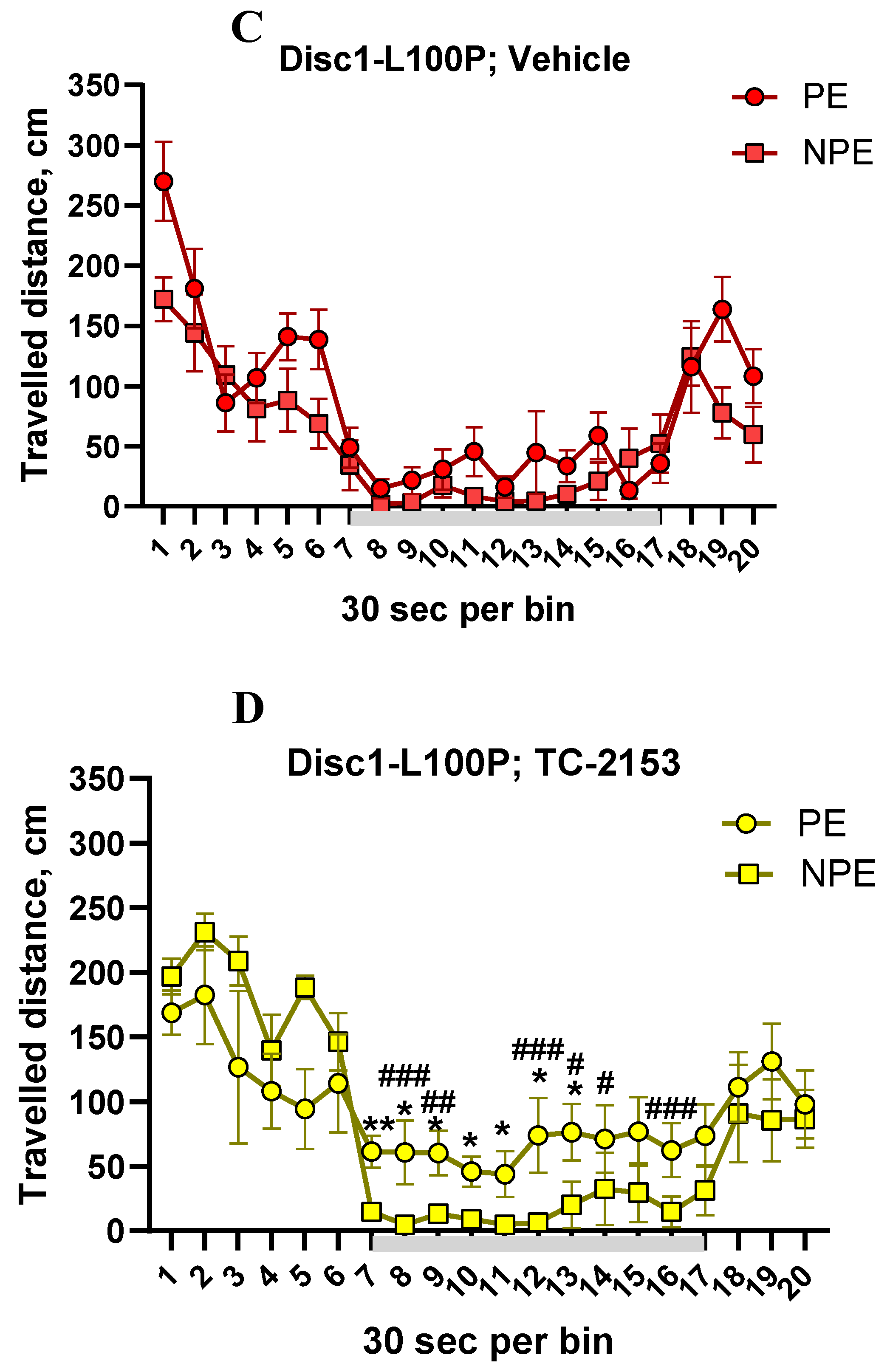

Table 1.
Effect of TC-2153 on the acoustic startle response in Disc1-L100P and WT mice.
| Group | WT | Disc1-L100P |
|---|---|---|
| Vehicle (n = 6-8) | 159.8 ± 34.9 | 90.0 ± 3.1 ### |
| TC-2153 (n = 6-8) | 132.4 ± 27.7 | 91.2 ± 5.7 |
### - p < 0.001 – in comparison with vehicle-treated WT mice.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



