Submitted:
18 July 2025
Posted:
21 July 2025
You are already at the latest version
Abstract
Twenty causative genes have been reported that cause non-syndromic childhood glaucoma associated with anterior segment dysgenesis. FOXC1, PAX6 and PITX2 are the most well-known, but cases linked to SLC4A11, PITX3 and SOX11 have also been reported. As genetic testing becomes increasingly widespread and rates of molecular diagnosis rise, the extent of phenotypic overlap between the different genetic causes of non-syndromic glaucoma associated with anterior segment dysgenesis is becoming more evident. Taking aniridia as an example, whilst PAX6 mutations remain the predominant cause, variants in CYP1B1, FOXC1, PXDN and SOX11 have also been reported in patients with childhood glaucoma and aniridia. Developments in molecular-based therapies for retinal and corneal disease are advancing rapidly, and pre-clinical studies of gene-based treatments for glaucoma and aniridia are showing promising results. Use of adeno-associated viral vectors for gene delivery is most common, with improvements in intraocular pressure and retinal ganglion cell survival in Tg-MYOCY437H mouse models of glaucoma, and successful correction of a germline PAX6G194X nonsense variant in mice using CRISPR/Cas9 gene editing. This review will explore the actions and interactions of the genetic causes of non-syndromic glaucoma associated with anterior segment dysgenesis and discuss current developments in molecular therapies for these patients.
Keywords:
childhood glaucoma
; anterior segment dysgenesis
; gene editing
; nonsense suppression
; aniridia
1. Introduction
Ocular development commences at gestational day 21 with induction of the eye field in the anterior neural plate prior to lateral evagination of the optic vesicles through the cephalic mesenchyme [1,2]. At gestational day 27, the optic vesicle contacts the surface ectoderm and signalling through bone morphogenetic protein (BMP) and retinoic acid first from the optic vesicle and then the lens placode stimulates invagination of both structures to form the optic cup and lens vesicle, which are present by day 37 (Figure 1a) [1]. By day 47, differentiation of the optic cup into primitive neural retina and retinal pigment epithelium (RPE) has commenced, and neural crest cells from the periocular mesenchyme begin to migrate into the developing anterior segment under the control of PAX6 (Figure 1b) [3]. PITX2 and FOXC1, expressed by the presumptive cornea, and PITX3 and FOXE3, expressed by the developing lens, have roles in regulating differentiation of the periocular mesenchyme into corneal keratocytes and endothelium, and detachment of the lens vesicle from the cornea, respectively (Figure 1c) [2]. From gestational week 15, PAX6, BMP4 and OTX1 stimulate elongation of the peripheral tips of the optic cup, which express CYP1B1 and MEIS2, along the anterior lens surface to form the epithelium of the ciliary body and iris (Figure 1d) [2,4,5,6]. Concurrently, a second wave of migration of the periocular mesenchyme into the anterior segment occurs, expressing PITX2, FOXC1 and FOXC2, which will form the ciliary body and iris stroma [2,3,5]. Trabecular meshwork development occurs from gestational week 20 with elongation, flattening and separation of mesenchymal cells in the iridocorneal angle to form lamellae, accompanied by vascular development in the adjacent sclera which becomes Schlemm’s canal (Figure 1e) [2,7].
Glaucoma is an overarching term for optic neuropathies resulting from retinal ganglion cell loss, which are typified by optic disc cupping and associated visual field defects. Childhood glaucoma, affecting individuals under 18 years of age, accounts for 5% of paediatric blindness worldwide [8]. In addition to optic nerve changes and visual field defects, features of childhood glaucoma include corneal enlargement and Haab striae, increased axial length and intraocular pressure >21mmHg; two or more features are required for diagnosis [9]. Childhood glaucoma is classified according to the underlying cause, and whether this is primary or secondary to another ocular or systemic condition [9]. Primary congenital glaucoma (Figure 2a) develops before 4 years of age and may be further divided into neonatal (0-1 month), infantile (>1-24 months) and late (>24 months) onset disease. It is a primary goniodysgenesis and presents with an immature appearance of the iridocorneal angle (Figure 2b) [9,10]. Juvenile open angle glaucoma presents after 4 years of age and with normal angle appearances. Secondary causes of childhood glaucoma include onset following cataract surgery, glaucoma associated with non-acquired ocular or systemic anomalies, and glaucoma associated with acquired conditions such as trauma and uveitis.
Anterior segment dysgenesis (ASD) is a broad term that incorporates developmental anomalies involving structures in the anterior segment of the eye [11,12,13]. These may affect a single structure, such as microcornea, or involve multiple structures, such as the iridocorneal and keratolenticular adhesions seen in Peters anomaly (Figure 2c). Between 30-75% of patients with ASD develop glaucoma depending on the subtype of dysgenesis [14,15], and young patients with either glaucoma or ASD should be thoroughly examined for features of each condition [13,15,16,17]. Many of the anterior segment dysgeneses have variable phenotypes with considerable overlap; for example, Axenfeld-Rieger anomaly (Figure 2d-e) may include corectopia, polycoria and iris hypoplasia in association with posterior embryotoxon, and like Peters anomaly, may also feature iridocorneal adhesions [15]. Patients with congenital aniridia (Figure 2f-h) present with underdevelopment of the iris of variable severity and foveal hypoplasia, although some will also have lenticular opacities and optic nerve hypoplasia from birth, many will develop glaucoma and aniridia-associated keratopathy by adulthood [14,18].
The molecular pathways involved in anterior segment development are highly complex and there is much overlap between genes that cause childhood glaucoma and those that cause anterior segment dysgenesis [11,12,13]. Many of the factors that contribute to this process remain unknown, and current molecular diagnostic rates for childhood glaucoma and anterior segment dysgenesis are below 25% [12,19], indicating the need for further studies to investigate additional genes and biological pathways that influence anterior segment development. This review summarises the genes identified to date in patients with non-syndromic childhood glaucoma associated with anterior segment dysgenesis, and discusses their functions, biological relationships and potential avenues for future treatments.
2. Results
Twenty genes linked to childhood glaucoma associated with anterior segment dysgenesis were identified, of which twelve were also reported to cause isolated childhood glaucoma. Reported phenotypes of anterior segment dysgenesis covered the full spectrum of cornea, iris, lens and mixed anomalies (Table 1). PAX6, FOXC1 and PITX2 are expressed in multiple anterior segment tissues during ocular development; other genes, such as CYP1B1, have more limited expression (Figure 3).
2.1. CYP1B1 (OMIM #601771)
CYP1B1 encodes a 543-amino acid enzyme which is part of the cytochrome P450 superfamily and is involved in retinol [56,57], melatonin [56] and 17ß-oestradiol metabolism [57,58]. It regulates myocilin production by limiting oestrogen-responsive activation of the MYOC promoter [58], and contributes to trabecular meshwork development through its modulation of periostin [59]. Pathogenic variants include missense, nonsense and frameshift mutations, and all have been identified throughout the gene [60]; the majority result in loss-of-function of metabolic activity [57,61,62], although some variants, such as c.1154C>A p.(Phe261Leu) and c.1776C>T p.(Arg469Trp) cause significantly increased retinol metabolism [57]. CYP1B1 is the most commonly identified genetic cause of primary congenital glaucoma (PCG) [60,63], causing 15-35% of PCG in European studies [12,61], and up to 100% of PCG in some ethnic groups [64]. Many CYP1B1 variants have also been associated with other anterior segment dysgeneses including aniridia [25], Axenfeld-Reiger anomaly [24] and Peters anomaly [20,21,22].
2.2. FOXC1 (OMIM #601090)
FOXC1, part of the forkhead box family of transcription factors, is expressed in the periocular mesenchyme and is necessary for separation of the cornea and lens in the developing eye [2,5,65]. It acts in conjunction with PITX2 to promote development of the corneal stroma and endothelium, iridocorneal angle and choroid, with co-expression of both transcription factors necessary for normal differentiation of these cellular populations [66]. In the trabecular meshwork, FOXC1 activity is mediated by miR-204 and regulates MEIS2 [67], a homeobox transcription factor which in turn is a direct regulator of PAX6 [68]. FOXC1 variants have been identified in patients with childhood glaucoma associated with Axenfeld-Rieger anomaly [15,23,29], Peters anomaly [13,28] and aniridia [13]. In the context of Axenfeld-Rieger syndrome, FOXC1 variants cause significantly more congenital glaucoma than do PITX2 variants, which are more associated with juvenile- and adult-onset glaucoma [15]. Whole gene deletions account for approximately one third of anterior segment dysgenesis caused by FOXC1 [12,15]. In a functional study, Medina-Trillo and colleagues showed that FOXC1 variants can result in either significantly increased or decreased transcriptional activity compared with wild-type; hypermorphic variants including c.141C>G p.(Tyr47*) and c.316C>T p.(Gln106*) were associated with isolated dominant glaucoma, whilst hypomorphic variants including c.377T>G p.(Ile126Ser) were identified in patients with Axenfeld-Rieger anomaly [69].
2.3. PAX6 (OMIM #607108)
Heterozygous pathogenic variants in PAX6 are typically associated with autosomal dominant aniridia, or are part of the Wilms tumour-aniridia-genital anomalies-retardation (WAGR) syndrome if the whole gene is deleted along with WT1 [11,18]. The PAX6 transcription factor is known to have a role in ocular development from gestational day 22 in the differentiation of the eye field and optic vesicle, through to the development of the optic cup and lens vesicle at day 37, and then in corneal, iris, ciliary body and trabecular meshwork development from day 47 onwards [2,70,71]. It has both paired and homeobox DNA-binding domains, and interacts with numerous other genes during this process, including SOX2, RAX, MEIS2 and ALDH1A3 [5,68,70]. Close regulation of PAX6 expression is required throughout embryogenesis to ensure normal development; under expression results in the aniridia spectrum of iris, foveal and optic nerve hypoplasia with associated glaucoma, cataracts and keratopathy, whilst overexpression causes microphthalmia, microcornea and ciliary body maldevelopment [72,73,74]. Whilst there are reports implicating PAX6 in isolated juvenile open angle glaucoma and childhood glaucoma associated with Peters anomaly, childhood glaucoma caused by PAX6 variants is very rarely diagnosed in patients without aniridia [28,75].
2.4. LTBP2 (OMIM #602091) and ADAMTSL4 (OMIM #610113)
Both LTBP2 and ADAMTSL4 contribute to cytoskeletal development through their interactions with fibrillin-1. LTBP2 encodes a 1821-amino acid extracellular matrix protein which interacts with fibrillin-1-containing microfibrils [76,77] and is expressed in the cornea, trabecular meshwork and lens [77]. A causative gene for Weill-Marchesani syndrome [23,31,76], which presents with short stature, brachydactyly, microspherophakia, ectopia lentis and myopia, LTBP2 has also been linked with congenital glaucoma associated with isolated microspherophakia [16], ectopia lentis [32] and congenital cataract [12]. The glycoprotein ADAMTSL4 binds fibrillin-1 and facilitates microfibril assembly, and is expressed in many ocular tissues including the cornea, iris and ciliary body in the anterior segment, and the retinal ganglion cells, retinal pigment epithelium, choroid and sclera in the posterior segment [78]. It has been linked to congenital glaucoma with ectopia lentis [33].
2.5. CPAMD8 (OMIM #608841)
CPAMD8 is expressed in the human eye from gestational week 9 onwards [79]. From weeks 9 through 22 of gestation, CPAMD8 expression increases in the lens and decreases in the retina, and strong expression is also observed in the iris and cornea at week 22 [79]. The pathways by which CPAMD8 acts to contribute to ocular development remain undefined, but recent work by Escribano and colleagues in zebrafish suggests an interaction between cpamd8 and adamtsl4 [80], implicating a potential contribution of CPAMD8 to development of fibrillar structures in the anterior segment. CPAMD8 variants have been linked to anterior segment dysgenesis associated with both congenital and juvenile glaucoma [16,34,35,36], and were identified in 3.7% of patients diagnosed with PCG in a large Australasian cohort [16].
2.6. COL4A1 (OMIM #120130) and PXDN (OMIM #605158)
Variants in COL4A1 and PXDN, which are both basement membrane components, have been identified in patients with anterior segment dysgeneses associated with childhood glaucoma [13,28,37,38,50,51]. COL4A1 encodes the alpha-1 subunit of type IV collagen, which is the main component of endothelial and epithelial basement membranes [81]. Peroxidasin, the protein product of the PXDN gene, forms sulfilimine crosslinks between type IV collagen molecules, and is necessary for normal development of type IV collagen-, laminin- and fibronectin-based fibrillar networks in endothelial cells [82]. Type IV collagens are found throughout the developing eye, and by embryonic day 18.5 (E18.5) the alpha-1 isoform can be identified in all anterior segment tissues in mice [83].
2.7. TEK (OMIM #600221)
One of two receptor tyrosine kinases in the angiopoietin/TEK signalling pathway, autosomal dominant mutations in TEK have been identified in patients with primary congenital glaucoma [16] and congenital glaucoma associated with sclerocornea [31]. In mice, TEK is expressed in Schlemm’s canal endothelial cells, and its ligand Angpt1 is expressed in the trabecular meshwork [84,85]. Supporting the role of TEK in iridocorneal angle and Schlemm’s canal development, studies of Angpt1 knockout mice have revealed severe hypoplasia of both Schlemm’s canal and the trabecular meshwork [85].
2.8. SLC4A11 (OMIM #610206)
Pathogenic variants in SLC4A11 are the most commonly identified genetic cause of congenital hereditary endothelial dystrophy (CHED) [86], which presents with early onset corneal oedema and, rarely, concomitant congenital glaucoma [16,27,41]. SLC4A11 localises to the corneal endothelium, where it contributes to osmotic homeostasis of the cornea and thus to maintaining corneal transparency [86,87]. Homozygous missense variants cause approximately half of SLC4A11-related disease [86], and both families with congenital glaucoma whose variants have been reported had homozygous missense mutations [27,41].
2.9. ADAMTS18 (OMIM #607512)
Part of the ADAMTS zinc-dependent metalloproteinase family, ADAMTS18 is expressed in adnexal, anterior segment and retinal cells during murine ocular development, with expression persisting postnatally in the lens and lacrimal gland [88,89]. It is a key factor in branching morphogenesis of the embryonic lacrimal gland [88], but its role in other aspects of ocular development remains unclear. Pathogenic variants in ADAMTS18 cause microcornea, myopic chorioretinal atrophy, and telecanthus (MMCAT) [90] and Knobloch syndrome (high myopia, vitreoretinal degeneration, retinal detachment, cataract and occipital encephalocoele) [91], but childhood glaucoma associated with microcornea has been reported in twins with compound heterozygous missense mutations of ADAMTS18 identified through panel testing [42].
2.10. PITX2 (OMIM #601542) and PITX3 (OMIM #602669)
The bicoid-type homeodomain transcription factors PITX2 and PITX3 have a role in embryonic anterior segment and lens development [92,93]. As discussed above, PITX2 interacts with FOXC1 and in mice they are co-expressed in the periocular mesenchyme from E11.5 [66]. It is therefore unsurprising that the ocular phenotypes of PITX2-related disease are similar to those of FOXC1 and include childhood glaucoma associated with Axenfeld-Rieger anomaly [16,28] and Peters anomaly [13,16]. PITX3 is expressed in the lens placode at murine E10 and continues to be expressed in the lens throughout development, in addition to expression in the midbrain, tongue and other embryological craniofacial tissues [93]. Variants have been identified in patients with congenital glaucoma associated with microcornea and congenital cataract [43,44].
2.11. FOXE3 (OMIM #601094)
FOXE3 variants are associated with a wide spectrum of anterior segment phenotypes and have been identified in patients with childhood glaucoma associated with Peters anomaly [45,46,47], congenital cataracts/aphakia [46,48] and microphthalmia [46,49]. The main role of the FOXE3 transcription factor during embryogenesis is in the development of the lens, where it is expressed in mice from E12.5 onwards and directly regulates the autophagy-associated protein DNAJB1 [45]. Functional studies of FOXE3 missense variants and small deletions isolated from patients with recessive disease showed significantly reduced transcriptional activity with homozygous or compound heterozygous variants, but no disease phenotype in their heterozygous parents, indicating that the presence of a single functional allele is sufficient for normal anterior segment development [48].
2.12. SOX11 (OMIM #600898)
The SOX11 SRY-Box transcription factor is a close homologue of SOX2 [94], which is known to play a key role in ocular development from induction of the eye field at gestational day 21 onwards [70]. During Xenopus embryogenesis sox11 is expressed in the optic vesicle, retina and anterior segment, and morpholino oligonucleotide (MO) knockdown of sox11 results in decreased pax6 expression in the eye field, loss of retinal lamination and a microphthalmic phenotype [95]. In Sox11 knockout mice, Pax6 expression is unaffected but BMP7 expression is reduced, and the phenotype includes lenticular defects, Peters anomaly and anterior ocular coloboma [96]. SOX11 variants have been reported in patients with congenital glaucoma associated with Peters anomaly and aniridia [31,52], although the gene is more commonly associated with intellectual developmental disorder with microcephaly with or without ocular malformations or hypogonadotropic hypogonadism (IDDMOH; Coffin Siris syndrome), of which congenital glaucoma can be a feature [23,52].
2.13. GJA8 (OMIM #600897)
GJA8 encodes connexin 50 (Cx50), a gap junction protein of lens fibre cells which is necessary for maintaining lens transparency and cellular homeostasis [23,97]. Studies of the missense mutation c.217T>C p.(Ser73Pro) in transfected human lens epithelial cells have shown aggregation of the abnormal protein in the cytosol with loss of localisation to the plasma membrane, resulting in reduced function of gap junction channels and increased apoptosis [97]. Although GJA8 variants are more commonly associated with congenital cataract [11], missense variants have also been identified in patients with congenital glaucoma associated with sclerocornea [23,28,31].
2.14. KERA (OMIM #603288)
KERA encodes keratocan, a keratan sulfate proteoglycan of the corneal stroma with a central role in maintaining collagen spacing and thus corneal transparency [98]. In adults, it is localised to stromal keratocytes [98]; during embryological development, studies of chicks by Conrad [99] and Gealy [100] have shown expression of Kera in the periocular mesenchyme from E4.5, with subsequent localisation to the anterior corneal stroma. Mutations in KERA are predominantly linked to cornea plana, in which there is flattening of the normal convex curvature of the cornea resulting in hyperopia [101], but one patient with anterior segment dysgenesis and congenital glaucoma associated with a homozygous nonsense KERA mutation has been reported [34].
2.15. CDH2 (OMIM #114020), KDM5C (OMIM #314690) and TFAP2A (OMIM #107580)
CDH2, KDM5C and TFAP2A are involved in neural development, with roles including axonal pathfinding [102], neuronal differentiation [103] and neural crest migration [104], respectively. All are reported to be causative for congenital glaucoma associated with Peters anomaly [53,54,55]. CDH2 encodes N-cadherin, which contributes to cell-cell adhesion in multiple tissues [53,102]. It is expressed in murine lens cells and the lens stalk at E10.75-11.0, and in lens and corneal endothelial cells at E17.5 [53]. Kdm5c is strongly expressed throughout Xenopus neural and ocular development, particularly in the lens and retina, and MO knockdown studies have confirmed the essential role of this histone demethylase in this process [105]. In human eyes, expression of the transcription factor TFAP2A is seen in the anterior lens epithelium from gestational day 35 and in the equatorial lens epithelium, secondary lens fibres and neural retinal by day 54 [106]. Whilst the detailed mechanism by which TFAP2A acts has not yet been elucidated, a study of embryological tooth development in mice showed dysregulation of Pitx2 and Sox2 in knockout models [107].
3. Management of Childhood Glaucoma and Anterior Segment Dysgenesis
Current treatment options for anterior segment dysgenesis and childhood glaucoma are predominantly surgical, although refractive, medical and supportive interventions, including genetic counselling and family planning, all contribute to the holistic management of these patients and their families [17,72,108]. For congenital glaucoma, angle surgeries (goniotomy and trabeculotomy) are first line; trabeculectomy with anti-fibrotic agents, glaucoma drainage devices and cyclodestructive procedures such as transscleral cyclophotocoagulation are used in refractory cases. Medical treatments (beta-blockers, carbonic anhydrase inhibitors and prostaglandin analogues) are used as adjuncts to surgery or as temporising measures while awaiting a procedure [108,109]. In patients with aniridia, cataract surgery and corneal and limbal grafts, alongside ocular lubricants, refraction and treatment of associated glaucoma have long been the mainstay of treatment [25,72]. Many patients with Peters anomaly require penetrating keratoplasty, although newer grafting techniques such as Descemet stripping automated endothelial keratoplasty (DSAEK) have been successfully employed in selected patients, and optical iridectomy may improve vision in patients with a smaller area of corneal opacity [17,110]. In all cases, refraction and amblyopia management should be provided as necessary, alongside educational and family support. Examination under anaesthetic should be considered for young children to enable detailed examination and identification of subtle signs of anterior segment dysgenesis or associated ocular conditions. Genetic testing is paramount to enable accurate genetic counselling, family planning advice and referral to other specialties in cases that may have an associated syndromic element. Furthermore, as the potential for molecular-based therapies is beginning to translate into practice with promising research including developments in gene editing and nonsense suppression drugs, genetic testing will allow patients access to targeted research.
4. Molecular Therapies Under Development
There are currently 41 FDA-approved cellular and gene therapies [111], of which only Luxturna (voretigene neparvovec-rzyl) targets an ocular condition, RPE65-related retinal dystrophy. Growing research into this field as a potential source of therapy for glaucoma and anterior segment disorders has focussed on animal and in vitro models, but in recent years this has begun translating into clinical trials [72,112]. Most of these studies have investigated treatments for corneal diseases and the various methods of gene delivery. Transduction of target cells with recombinant adenoviruses (rAds) and adeno-associated viruses (rAAVs) are the most common viral vectors [112,113,114], although retroviruses [115] and lentiviruses [116] have been used in some studies. Non-viral vectors include metal and magnetic nanoparticles, micelles and lipid nanoparticles [112,117,118,119], which may be delivered topically or by injection. Alternative methods that do not require a vector include corneal electroporation and iontophoresis [120,121]. In the anterior segment, research into gene-based therapies has focused predominantly on corneal and ocular surface disease, adult-onset glaucoma and aniridia [72,112,122,123], with no published trials of gene therapies for Peters anomaly or Axenfeld-Rieger syndrome. Here we summarise current research into cellular and gene-based therapies for glaucoma and aniridia.
4.1. Gene-Based Therapies for Glaucoma
Molecular-based therapies under development for glaucoma are predominantly targeted towards modification of the aqueous production/outflow pathways and minimising damage to retinal ganglion cells (RGCs) [122], as these are key factors in the development of multifactorial adult-onset glaucoma. Sulak and colleagues [123] identified 153 studies into gene-based therapies for glaucoma and noted that there were twice as many therapies targeting RGC neuroprotection compared with those targeting aqueous production and outflow. Strategies to modulate the molecular pathways involved in aqueous regulation are in development, with targets including myocilin (MYOC), aquaporin-1 (AQP1) and matrix metalloproteinases (MMPs).
Mutations in the MYOC gene, encoding the extracellular matrix protein myocilin, are the most commonly identified single gene cause of primary open angle glaucoma (POAG) and juvenile open angle glaucoma (JOAG), identified in 2-4% and 10-30% of cases, respectively [122]. Dominant gain-of-function mutations result in intracellular accumulation of the abnormal protein, resulting in increasing IOP and resultant glaucomatous damage to the retinal ganglion cells and optic nerve [114]. Mice with the MYOC p.(Tyr437His) variant (Tg-MYOCY437H) can be used as a model for POAG, developing these same pathogenic features, and may be employed for in vivo confirmation of therapeutic benefit [114,116].
Gene editing approaches targeted to specific myocilin mutations are in development. Jain [114] used a CRISPR-Cas9 approach with an adenoviral Ad5 vector to introduce a truncating frameshift mutation into Tg-MYOCY437H mice. They demonstrated significant reduction of IOP into the normal range in both younger (age ≤1 month) and older (age ≥9 months) mice, with results sustained for at least 2 months post-injection. The group have subsequently shown positive results using the same CRISPR-Cas9 system with a lentiviral vector [116], with IOP reduction into the normal range in Tg-MYOCY437H mice maintained for at least 6 weeks post-injection. They additionally tested an AAV2 vector in vitro on human TM3 cells with Y437H or G364V MYOC mutations, but the reduction in accumulated myocilin was only 34% lower than untreated levels with the AAV2 vector compared with 62% reduction for the lentivirus. Another mechanism that has been proposed for reducing the pathogenicity of MYOC mutations is to use small interfering RNAs (siRNAs) to disrupt the expression of the gene. Preliminary results in TM5 cells demonstrated significant suppression of both wild type and mutant myocilin expression to 60% of control levels [124]. Whilst further work is needed, these approaches hold promise for potential gene-targeted therapies for MYOC-related glaucoma to become available in the future.
Another CRISPR-based approach employing a lentivirus vector carrying single guide RNA (sgRNA) to target the TGFβ2 promoter has been developed by Mao’s group [125]. Administration of the sgRNA lentivirus 2.5 weeks prior to induction of ocular hypertension prevented IOP increases, maintaining an IOP level below 20mmHg for at least 8 weeks post-injection, compared with an increase to over 30mmHg in controls. Aquaporin 1 (AQP1) has also been targeted with sgRNA. This transmembrane water channel is present in many tissues including the ciliary body, and deletion of Aqp1 has been shown to significantly reduce IOP in mice [126]. Introduction of two AQP1-indel-inducing sgRNAs within a single AAV vector given by intravitreal injection has been shown to transduce the murine ciliary body and reduce IOP by 22% after 3 weeks, with neuroprotective effects sustained to at least 7 weeks post-injection [127]. There was also effective transfection of ex vivo human ciliary body epithelial cells. AQP1 has therefore been proposed as a potential therapeutic target for glaucoma.
MMP activity is known to be reduced in glaucoma, and one of the key actions of the prostaglandin analogue drops used to treat open angle glaucoma is to increase MMP expression in the ciliary body [128]. Using an AAV9 vector to transduce corneal endothelial cells, O’Callaghan [129] introduced tetracycline-inducible murine MMP3 into Tg-MYOCY437H and dexamethasone-induced ocular hypertensive mice. In both groups, IOP 2 weeks after doxycycline induction was significantly reduced in the treated mice compared with controls. They also demonstrated significant increases in MMP3 expression and aqueous outflow in non-human primates in vivo, and increased aqueous outflow in human donor anterior segments in vitro using this approach [129]. Similar effects have been achieved using intracameral IκBα-silencing siRNA to transfect the anterior segment of nonhuman primates, with upregulation of MMP2 and MMP9 and resultant 49% reduction in IOP [130]. A lesser IOP reduction has been achieved through the use of the microRNA miR-21-5p, which is predicted to have downstream effects including activation of MMP9. Whilst preliminary results show potential, the maximal IOP reduction following administration was 17.8% [131], lower than the 20-35% achieved by current topical treatments [132].
Approaches to disrupt β2-adrenoceptors have also shown promise. Loma [132] topically administered siRNAs designed to silence β2-adrenoceptors to rabbits and compared the effects to commercially available topical dorzolamide, latanoprost and timolol. The siRNAs significantly reduced IOP by 30 ± 5% compared to controls, with effects detectable 24hrs after instillation and sustained for 5 days. The level of IOP reduction was comparable to that achieved by all commercial agents, but with a longer onset and offset time. A commercial compound, bamosiran, has recently completed its phase II trial. This RNA interference approach designed to prevent β2-adrenoceptor production showed non-inferiority to timolol in patients with a baseline IOP ≥25mmHg, but not across all patient groups [133]. However, the positive results achieved overall by these diverse approaches to molecular modulation of aqueous regulation lay a strong foundation for molecular therapies to become available for patients within the near future.
4.2. Gene-Based Therapies for Aniridia
A novel approach to treatment of aniridia is nonsense suppression therapy, using drugs that impair recognition of premature termination codons (PTCs) during translation to allow the production of a full-length protein [134]. In aniridia patients in whom a genetic cause is identified, dominant pathogenic PAX6 mutations are responsible for approximately 90% of cases [14]; of these, nonsense and frameshift mutations resulting in a PTC comprise 70% of pathogenic and likely pathogenic PAX6 variants [135], so a treatment that targets these mutations has the potential to benefit a large proportion of these patients. Promising results using nonsense suppression therapies, which work on in-frame nonsense mutations, have been reported in vitro [136] and in the Sey mouse model [137,138]. Sey mice with a heterozygous PAX6 p.(Gly194X) nonsense mutation treated with topical nonsense suppression drugs from postnatal day 14-60 developed partial phenotypic rescue, including reversal of corneal, lens and retinal abnormalities [137,138]. However, these benefits have not yet been translated into positive results in clinical trials, with the Study of Ataluren in Participants With Nonsense Mutation Aniridia (STAR) unfortunately failing to meet its primary endpoints [139].
Gene editing is under investigation as another potential avenue of treatment for aniridia. Work in Sey mice has shown successful germline correction of the PAX6 p.(Gly194X) mutation in vivo by microinjection of the targeted CRISPR/Cas9 complex into murine zygotes at day 0.5 post-coitus, with inheritance of the corrected gene by the offspring of these transgenic mice [140]. The progeny had normal eye development and normal PAX6 levels in neural tissue at embryonic day 18.5, when microphthalmic changes are observed in Sey mice [140]. Further work has demonstrated the potential use of AAV vectors for delivery of gene therapy in aniridia, with results suggesting that the AAV9 capsid is able to safely transduce corneal and retinal cells of Sey mice [113].
Work to expand the therapeutic options for management of aniridia-associated keratopathy has shown that duloxetine and ritanserin, both anti-serotonin drugs which inhibit the MEK/ERK signalling pathway, can enhance endogenous PAX6 expression in haploinsufficient corneal limbal epithelial stem cells [141–143]. A regulatory microRNA, miR-204-5p, has also been reported that upregulates PAX6 in PAX6-knockdown corneal limbal stem cells and mature differentiated corneal epithelial cells in vitro; there was no significant change in PAX6 expression in primary human limbal epithelial cells, human limbal stem cells or in the corneas of Pax6(Sey/+) mice transfected with miR-204-5p [144]. Further work is needed to determine the effects of these modulating factors in other ocular cell types, but they provide another potential avenue for therapeutic development. However, any strategies to increase PAX6 expression during embryogenesis need to ensure that induced levels remain carefully modulated. Both under- and overexpression of PAX6 can lead to ocular developmental anomalies as discussed earlier [73,74], therefore ensuring corrected PAX6 expression levels are as close as possible to the normal embryological state is essential when correcting pathogenic variants. To address this, Simpson’s team [145] have designed PAX6 MiniPromoters and reported their robust and specific expression within the PAX6-positive RGC, amacrine, horizontal and Müller glial cells, with significantly better regulation of PAX6 expression than achieved by the ubiquitous smCBA promoter.
4. Methods
A combination of strategies was used to identify genes that cause non-syndromic childhood glaucoma associated with anterior segment dysgenesis. A list was compiled of all green and amber genes in the NHS Genomics Medicine Service’s “Structural Eye Disease” panel (v4.1) and all genes and conditions associated with the term “glaucoma” in Online Mendelian Inheritance in Man (OMIM) [146]. Each gene identified was then searched on PubMed for reports and studies confirming its association with childhood glaucoma and any anterior segment developmental anomaly. Any additional genes identified through these publications which met the criteria were also reviewed. Genes identified in patients with syndromic ASD, or in whom glaucoma was identified subsequently to cataract surgery, were excluded.
5. Conclusions
As the accessibility of genetic testing for ocular diseases has increased, so too has the number of genes identified that are linked to childhood glaucoma associated with anterior segment dysgenesis, although molecular diagnostic rates for these conditions remain low. To date, twenty genes have been associated with this subset of ocular developmental disorders, with many more involved in the pathogenesis of primary congenital glaucoma and anterior segment dysgenesis without glaucoma [11]. The pathways governing anterior segment development are complex and mutations in different genes can cause very similar clinical features. Furthermore, there is significant variation in the phenotype resulting from mutations within individual genes. Improvements in individualised patient prognostication and management will benefit from anatomical-based reporting of clinical features and gene-based diagnosis, both for identifying the most appropriate therapies and allowing detailed genotype-phenotype correlation studies of these rare conditions in the future. Additionally, it is a step towards the change in approach to treatment required in this advent of gene therapy. As the development of these targeted treatments begins to translate into clinically available management options, knowing a patient’s underlying genetic diagnosis will be key to ensuring they receive appropriate treatment.
Author Contributions
Conceptualization, N.C. and M.M.; methodology, N.C.; writing—original draft preparation, N.C.; writing—review and editing, C.M. and M.M.; visualization, N.C.; supervision, M.M. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by Moorfields Eye Charity, the Wellcome Trust (205174/Z/16/Z), the National Institute for Health and Care Research (NIHR), Biomedical Research Centre at Moorfields Eye Hospital NHS Foundation Trust, and UCL Institute of Ophthalmology.
Data Availability Statement
No new data were created or analyzed in this study. Data sharing is not applicable to this article.
Conflicts of Interest
The authors declare no conflicts of interest.
Abbreviations
The following abbreviations are used in this manuscript:
| ASD | Anterior segment dysgenesis |
| BMP | Bone morphogenetic protein |
| CHED | Congenital hereditary endothelial dystrophy |
| CRISPR/Cas9 | Clustered regularly interspaced palindromic repeats/CRISPR-associated protein 9 |
| DSAEK | Descemet stripping automated endothelial keratoplasty |
| ECM | Extracellular matrix |
| FDA | US Food and Drug Administration |
| IOP | Intraocular pressure |
| JOAG | Juvenile open angle glaucoma |
| MEK/ERK | Mitogen-activated protein kinase-ERK kinase/extracellular signal related kinase |
| MMCAT | Microcornea, myopic chorioretinal atrophy and telecanthus |
| MMP | Matrix metalloproteinase |
| MO | Morpholino oligonucleotide |
| OMIM | Online Mendelian Inheritance in Man |
| PCG | Primary congenital glaucoma |
| POAG | Primary open angle glaucoma |
| PTC | Premature termination codon |
| rAAV | Recombinant adeno-associated virus |
| rAd | Recombinant adenovirus |
| sgRNA | Single guide RNA |
| siRNA | Small interfering RNA |
| RGC | Retinal ganglion cell |
| RPE | Retinal pigment epithelium |
| STAR | Study of Ataluren in Participants With Nonsense Mutation Aniridia |
References
- Shaham, O.; Menuchin, Y.; Farhy, C.; Ashery-Padan, R. Pax6: A Multi-Level Regulator of Ocular Development. Prog. Retin. Eye Res. 2012, 31, 351–376. [Google Scholar] [CrossRef] [PubMed]
- Cvekl, A.; Tamm, E.R. Anterior Eye Development and Ocular Mesenchyme: New Insights from Mouse Models and Human Diseases. BioEssays News Rev. Mol. Cell. Dev. Biol. 2004, 26, 374. [Google Scholar] [CrossRef] [PubMed]
- Takamiya, M.; Stegmaier, J.; Kobitski, A.Y.; Schott, B.; Weger, B.D.; Margariti, D.; Cereceda Delgado, A.R.; Gourain, V.; Scherr, T.; Yang, L.; et al. Pax6 Organizes the Anterior Eye Segment by Guiding Two Distinct Neural Crest Waves. PLoS Genet. 2020, 16, e1008774. [Google Scholar] [CrossRef] [PubMed]
- Stoilov, I.; Rezaie, T.; Jansson, I.; Schenkman, J.B.; Sarfarazi, M. Expression of Cytochrome P4501b1 (Cyp1b1) during Early Murine Development. Mol. Vis. 2004, 10, 629–636. [Google Scholar] [PubMed]
- Davis-Silberman, N.; Ashery-Padan, R. Iris Development in Vertebrates; Genetic and Molecular Considerations. Brain Res. 2008, 1192, 17–28. [Google Scholar] [CrossRef] [PubMed]
- Miesfeld, J.B.; Brown, N.L. Eye Organogenesis: A Hierarchical View of Ocular Development. Curr. Top. Dev. Biol. 2019, 132, 351–393. [Google Scholar] [CrossRef] [PubMed]
- Mizokami, K.; Sugiura, T.; San Juan, R.G. The Development of Human Trabecular Meshwork. Med. Electron Microsc. 1994, 27, 275–281. [Google Scholar] [CrossRef]
- Marchini, G.; Toscani, M.; Chemello, F. Pediatric Glaucoma: Current Perspectives. Pediatr. Health Med. Ther. 2014, 5, 15–27. [Google Scholar] [CrossRef]
- Thau, A.; Lloyd, M.; Freedman, S.; Beck, A.; Grajewski, A.; Levin, A.V. New Classification System for Pediatric Glaucoma: Implications for Clinical Care and a Research Registry. Curr. Opin. Ophthalmol. 2018, 29, 385–394. [Google Scholar] [CrossRef] [PubMed]
- Ko, F.; Papadopoulos, M.; Khaw, P.T. Primary Congenital Glaucoma. Prog. Brain Res. 2015, 221, 177–189. [Google Scholar] [CrossRef] [PubMed]
- Reis, L.M.; Seese, S.; Costakos, D.; Semina, E.V. Congenital Anterior Segment Ocular Disorders: Genotype-Phenotype Correlations and Emerging Novel Mechanisms. Prog. Retin. Eye Res. 2024, 101288. [Google Scholar] [CrossRef] [PubMed]
- Patel, A.; Hayward, J.D.; Tailor, V.; Nyanhete, R.; Ahlfors, H.; Gabriel, C.; Jannini, T.B.; Abbou-Rayyah, Y.; Henderson, R.; Nischal, K.K.; et al. The Oculome Panel Test: Next-Generation Sequencing to Diagnose a Diverse Range of Genetic Developmental Eye Disorders. Ophthalmology 2019, 126, 888–907. [Google Scholar] [CrossRef] [PubMed]
- Reis, L.M.; Semina, E.V. Genetics of Anterior Segment Dysgenesis Disorders. Curr. Opin. Ophthalmol. 2011, 22, 314–324. [Google Scholar] [CrossRef] [PubMed]
- Hingorani, M.; Hanson, I.; van Heyningen, V. Aniridia. Eur. J. Hum. Genet. 2012, 20, 1011–1017. [Google Scholar] [CrossRef] [PubMed]
- Reis, L.M.; Maheshwari, M.; Capasso, J.; Atilla, H.; Dudakova, L.; Thompson, S.; Zitano, L.; Lay-Son, G.; Lowry, R.B.; Black, J.; et al. Axenfeld-Rieger Syndrome: More than Meets the Eye. J. Med. Genet. 2023, 60, 368–379. [Google Scholar] [CrossRef] [PubMed]
- Knight, L.S.W.; Ruddle, J.B.; Taranath, D.A.; Goldberg, I.; Smith, J.E.H.; Gole, G.; Chiang, M.Y.; Willett, F.; D’Mellow, G.; Breen, J.; et al. Childhood and Early Onset Glaucoma Classification and Genetic Profile in a Large Australasian Disease Registry. Ophthalmology 2021, 128, 1549–1560. [Google Scholar] [CrossRef] [PubMed]
- Wowra, B.; Dobrowolski, D.; Parekh, M.; Wylęgała, E. General Treatment and Ophthalmic Management of Peters’ Anomaly. J. Clin. Med. 2024, 13, 532. [Google Scholar] [CrossRef] [PubMed]
- Moosajee, M.; Hingorani, M.; Moore, A.T. PAX6-Related Aniridia. In GeneReviews®; Adam, M.P., Feldman, J., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Amemiya, A., Eds.; University of Washington, Seattle: Seattle (WA), 1993. [Google Scholar]
- Jackson, D.; Malka, S.; Harding, P.; Palma, J.; Dunbar, H.; Moosajee, M. Molecular Diagnostic Challenges for Non-retinal Developmental Eye Disorders in the United Kingdom. Am. J. Med. Genet. C Semin. Med. Genet. 2020, 184, 578–589. [Google Scholar] [CrossRef] [PubMed]
- Home - OMIM. Available online: https://omim.org/ (accessed on 1 October 2024).
- Vincent, A.; Billingsley, G.; Priston, M.; Glaser, T.; Oliver, E.; Walter, M.; Ritch, R.; Levin, A.; Heon, E. Further Support of the Role of CYP1B1 in Patients with Peters Anomaly. Mol. Vis. 2006, 12, 506–510. [Google Scholar] [PubMed]
- Edward, D.; Rajhi, A.A.; Lewis, R.A.; Curry, S.; Wang, Z.; Bejjani, B. Molecular Basis of Peters Anomaly in Saudi Arabia. Ophthalmic Genet. 2004, 25, 257–270. [Google Scholar] [CrossRef] [PubMed]
- Churchill, A.J.; Yeung, A. A Compound Heterozygous Change Found in Peters’ Anomaly. Mol. Vis. 2005, 11, 66–70. [Google Scholar] [PubMed]
- Stingl, J.V.; Diederich, S.; Diel, H.; Schuster, A.K.; Wagner, F.M.; Chronopoulos, P.; Aghayeva, F.; Grehn, F.; Winter, J.; Schweiger, S.; et al. First Results from the Prospective German Registry for Childhood Glaucoma: Phenotype-Genotype Association. J. Clin. Med. 2021, 11, 16. [Google Scholar] [CrossRef] [PubMed]
- Tanwar, M.; Dada, T.; Dada, R. Axenfeld-Rieger Syndrome Associated with Congenital Glaucoma and Cytochrome P4501B1 Gene Mutations. Case Rep. Med. 2010, 2010, 212656. [Google Scholar] [CrossRef] [PubMed]
- Samant, M.; Chauhan, B.K.; Lathrop, K.L.; Nischal, K.K. Congenital Aniridia: Etiology, Manifestations and Management. Expert Rev. Ophthalmol. 2016, 11, 135–144. [Google Scholar] [CrossRef] [PubMed]
- Franco, E.; Gagrani, M.; Scanga, H.L.; Areaux, R.G.; Chu, C.T.; Nischal, K.K. Variable Phenotype of Congenital Corneal Opacities in Biallelic CYP1B1 Pathogenic Variants. Cornea 2024, 43, 195–200. [Google Scholar] [CrossRef] [PubMed]
- Al-Saei, O.; Malka, S.; Owen, N.; Aliyev, E.; Vempalli, F.R.; Ocieczek, P.; Al-Khathlan, B.; Genomics England Research Consortium; Fakhro, K.; Moosajee, M. Increasing the Diagnostic Yield of Childhood Glaucoma Cases Recruited into the 100,000 Genomes Project. BMC Genomics 2024, 25, 484. [Google Scholar] [CrossRef] [PubMed]
- Ma, A.; Yousoof, S.; Grigg, J.R.; Flaherty, M.; Minoche, A.E.; Cowley, M.J.; Nash, B.M.; Ho, G.; Gayagay, T.; Lai, T.; et al. Revealing Hidden Genetic Diagnoses in the Ocular Anterior Segment Disorders. Genet. Med. Off. J. Am. Coll. Med. Genet. 2020, 22, 1623–1632. [Google Scholar] [CrossRef] [PubMed]
- Thanikachalam, S.; Hodapp, E.; Chang, T.C.; Swols, D.M.; Cengiz, F.B.; Guo, S.; Zafeer, M.F.; Seyhan, S.; Bademci, G.; Scott, W.K.; et al. Spectrum of Genetic Variants Associated with Anterior Segment Dysgenesis in South Florida. Genes 2020, 11, 350. [Google Scholar] [CrossRef] [PubMed]
- Collantes, E.R.A.; Delfin, M.S.; Fan, B.; Torregosa, J.M.R.; Siguan-Bell, C.; de Guzman Florcruz, N.V.; Martinez, J.M.D.; Masna-Hidalgo, B.J.; Guzman, V.P.T.; Anotado-Flores, J.F.; et al. EFEMP1 Rare Variants Cause Familial Juvenile-Onset Open Angle Glaucoma. Hum. Mutat. 2022, 43, 240–252. [Google Scholar] [CrossRef] [PubMed]
- Aghayeva, F.A.; Schuster, A.K.; Diel, H.; Chronopoulos, P.; Wagner, F.M.; Grehn, F.; Pirlich, N.; Schweiger, S.; Pfeiffer, N.; Hoffmann, E.M. Childhood Glaucoma Registry in Germany: Initial Database, Clinical Care and Research (Pilot Study). BMC Res. Notes 2022, 15, 32. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Xu, T.; Gan, J.; Mao, Y.; Zhao, L.; Jiao, X.; Fan, M.; Wang, T.; Zhang, D.; Xu, M.; et al. Zonule-Associated Gene Variants in Isolated Ectopia Lentis and Glaucoma. J. Glaucoma 2023, 32, e80–e89. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.-X.; Jia, W.-N.; Sun, Y.; Chen, T.-H.; Zhao, Z.-N.; Lan, L.-N.; Liu, Y.; Song, L.-H.; Jiang, Y.-X. Biallelic ADAMTSL4 Variants in a Chinese Cohort of Congenital Ectopia Lentis: Implications for Genotype-Phenotype Relationships. Hum. Mutat. 2022, 43, 2141–2152. [Google Scholar] [CrossRef] [PubMed]
- Alsaif, H.S.; Khan, A.O.; Patel, N.; Alkuraya, H.; Hashem, M.; Abdulwahab, F.; Ibrahim, N.; Aldahmesh, M.A.; Alkuraya, F.S. Congenital Glaucoma and CYP1B1: An Old Story Revisited. Hum. Genet. 2019, 138, 1043–1049. [Google Scholar] [CrossRef] [PubMed]
- Siggs, O.M.; Souzeau, E.; Taranath, D.A.; Dubowsky, A.; Chappell, A.; Zhou, T.; Javadiyan, S.; Nicholl, J.; Kearns, L.S.; Staffieri, S.E.; et al. Biallelic CPAMD8 Variants Are a Frequent Cause of Childhood and Juvenile Open-Angle Glaucoma. Ophthalmology 2020, 127, 758–766. [Google Scholar] [CrossRef] [PubMed]
- Bonet-Fernández, J.-M.; Aroca-Aguilar, J.-D.; Corton, M.; Ramírez, A.-I.; Alexandre-Moreno, S.; García-Antón, M.-T.; Salazar, J.-J.; Ferre-Fernández, J.-J.; Atienzar-Aroca, R.; Villaverde, C.; et al. CPAMD8 Loss-of-Function Underlies Non-Dominant Congenital Glaucoma with Variable Anterior Segment Dysgenesis and Abnormal Extracellular Matrix. Hum. Genet. 2020, 139, 1209–1231. [Google Scholar] [CrossRef] [PubMed]
- Zhu, A.Y.; Costain, G.; Cytrynbaum, C.; Weksberg, R.; Cohn, R.D.; Ali, A. Novel Heterozygous Variants in PXDN Cause Different Anterior Segment Dysgenesis Phenotypes in Monozygotic Twins. Ophthalmic Genet. 2021, 42, 624–630. [Google Scholar] [CrossRef] [PubMed]
- Khan, K.; Rudkin, A.; Parry, D.A.; Burdon, K.P.; McKibbin, M.; Logan, C.V.; Abdelhamed, Z.I.A.; Muecke, J.S.; Fernandez-Fuentes, N.; Laurie, K.J.; et al. Homozygous Mutations in PXDN Cause Congenital Cataract, Corneal Opacity, and Developmental Glaucoma. Am. J. Hum. Genet. 2011, 89, 464–473. [Google Scholar] [CrossRef] [PubMed]
- Zazo-Seco, C.; Plaisancié, J.; Bitoun, P.; Corton, M.; Arteche, A.; Ayuso, C.; Schneider, A.; Zafeiropoulou, D.; Gilissen, C.; Roche, O.; et al. Novel PXDN Biallelic Variants in Patients with Microphthalmia and Anterior Segment Dysgenesis. J. Hum. Genet. 2020, 65, 487–491. [Google Scholar] [CrossRef] [PubMed]
- Choi, A.; Lao, R.; Ling-Fung Tang, P.; Wan, E.; Mayer, W.; Bardakjian, T.; Shaw, G.M.; Kwok, P.-Y.; Schneider, A.; Slavotinek, A. Novel Mutations in PXDN Cause Microphthalmia and Anterior Segment Dysgenesis. Eur. J. Hum. Genet. EJHG 2015, 23, 337–341. [Google Scholar] [CrossRef] [PubMed]
- Yousaf, K.; Naz, S.; Mushtaq, A.; Wohler, E.; Sobreira, N.; Ho, B.-M.; Chen, L.-J.; Chu, W.-K.; Bashir, R. Exome Sequencing Reveals SLC4A11 Variant Underlying Congenital Hereditary Endothelial Dystrophy (CHED2) Misdiagnosed as Congenital Glaucoma. Genes 2023, 14, 310. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.Y.; Tao, Y.F.; Mao, Y.K.; Chen, Z.J.; Wang, Y.; Hong, Y.F.; Fan, N. A family with developmental glaucoma and microcornea due to novel ADAMTS18 gene mutations. Zhonghua Yan Ke Za Zhi Chin. J. Ophthalmol. 2024, 60, 78–83. [Google Scholar] [CrossRef]
- Zhou, L.; Xu, Z.; Wu, Q.; Wei, X. Unilateral Buphthalmos, Corneal Staphyloma and Corneal Fistula Caused by Pathogenic Variant in the PITX3 Gene: A Case Report. BMC Ophthalmol. 2022, 22, 385. [Google Scholar] [CrossRef] [PubMed]
- Verdin, H.; Sorokina, E.A.; Meire, F.; Casteels, I.; de Ravel, T.; Semina, E.V.; De Baere, E. Novel and Recurrent PITX3 Mutations in Belgian Families with Autosomal Dominant Congenital Cataract and Anterior Segment Dysgenesis Have Similar Phenotypic and Functional Characteristics. Orphanet J. Rare Dis. 2014, 9, 26. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.Y.; Vasanth, S.; Kabir, F.; Gottsch, J.D.; Khan, A.O.; Chaerkady, R.; Lee, M.-C.W.; Leitch, C.C.; Ma, Z.; Laux, J.; et al. FOXE3 Contributes to Peters Anomaly through Transcriptional Regulation of an Autophagy-Associated Protein Termed DNAJB1. Nat. Commun. 2016, 7, 10953. [Google Scholar] [CrossRef] [PubMed]
- Plaisancié, J.; Ragge, N.K.; Dollfus, H.; Kaplan, J.; Lehalle, D.; Francannet, C.; Morin, G.; Colineaux, H.; Calvas, P.; Chassaing, N. FOXE3 Mutations: Genotype-Phenotype Correlations. Clin. Genet. 2018, 93, 837–845. [Google Scholar] [CrossRef] [PubMed]
- Ormestad, M.; Blixt, A.; Churchill, A.; Martinsson, T.; Enerbäck, S.; Carlsson, P. Foxe3 Haploinsufficiency in Mice: A Model for Peters’ Anomaly. Invest. Ophthalmol. Vis. Sci. 2002, 43, 1350–1357. [Google Scholar] [PubMed]
- Islam, L.; Kelberman, D.; Williamson, L.; Lewis, N.; Glindzicz, M.B.; Nischal, K.K.; Sowden, J.C. Functional Analysis of FOXE3 Mutations Causing Dominant and Recessive Ocular Anterior Segment Disease. Hum. Mutat. 2015, 36, 296–300. [Google Scholar] [CrossRef] [PubMed]
- Rashid, M.; Qasim, M.; Ishaq, R.; Bukhari, S.A.; Sajid, Z.; Ashfaq, U.A.; Haque, A.; Ahmed, Z.M. Pathogenic Variants of AIPL1, MERTK, GUCY2D, and FOXE3 in Pakistani Families with Clinically Heterogeneous Eye Diseases. PLoS ONE 2020, 15, e0239748. [Google Scholar] [CrossRef] [PubMed]
- Sibon, I.; Coupry, I.; Menegon, P.; Bouchet, J.-P.; Gorry, P.; Burgelin, I.; Calvas, P.; Orignac, I.; Dousset, V.; Lacombe, D.; et al. COL4A1 Mutation in Axenfeld-Rieger Anomaly with Leukoencephalopathy and Stroke. Ann. Neurol. 2007, 62, 177–184. [Google Scholar] [CrossRef] [PubMed]
- Coupry, I.; Sibon, I.; Mortemousque, B.; Rouanet, F.; Mine, M.; Goizet, C. Ophthalmological Features Associated with COL4A1 Mutations. Arch. Ophthalmol. Chic. Ill 1960 2010, 128, 483–489. [Google Scholar] [CrossRef] [PubMed]
- Diel, H.; Ding, C.; Grehn, F.; Chronopoulos, P.; Bartsch, O.; Hoffmann, E.M. First Observation of Secondary Childhood Glaucoma in Coffin-Siris Syndrome: A Case Report and Literature Review. BMC Ophthalmol. 2021, 21, 28. [Google Scholar] [CrossRef] [PubMed]
- Reis, L.M.; Houssin, N.S.; Zamora, C.; Abdul-Rahman, O.; Kalish, J.M.; Zackai, E.H.; Plageman, T.F.; Semina, E.V. Novel Variants in CDH2 Are Associated with a New Syndrome Including Peters Anomaly. Clin. Genet. 2020, 97, 502–508. [Google Scholar] [CrossRef] [PubMed]
- Reis, L.M.; Atilla, H.; Kannu, P.; Schneider, A.; Thompson, S.; Bardakjian, T.; Semina, E.V. Distinct Roles of Histone Lysine Demethylases and Methyltransferases in Developmental Eye Disease. Genes 2023, 14, 216. [Google Scholar] [CrossRef] [PubMed]
- Weh, E.; Reis, L.M.; Happ, H.C.; Levin, A.V.; Wheeler, P.G.; David, K.L.; Carney, E.; Angle, B.; Hauser, N.; Semina, E.V. Whole Exome Sequence Analysis of Peters Anomaly. Hum. Genet. 2014, 133, 1497–1511. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.; Iwata, T. Exploring the Genetic Landscape of Childhood Glaucoma. Child. Basel Switz. 2024, 11, 454. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, A.; Chakraborty, S.; Chakraborty, A.; Chakrabarti, S.; Ray, K. Functional and Structural Analyses of CYP1B1 Variants Linked to Congenital and Adult-Onset Glaucoma to Investigate the Molecular Basis of These Diseases. PloS One 2016, 11, e0156252. [Google Scholar] [CrossRef] [PubMed]
- Mookherjee, S.; Acharya, M.; Banerjee, D.; Bhattacharjee, A.; Ray, K. Molecular Basis for Involvement of CYP1B1 in MYOC Upregulation and Its Potential Implication in Glaucoma Pathogenesis. PloS One 2012, 7, e45077. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Wang, S.; Sorenson, C.M.; Teixeira, L.; Dubielzig, R.R.; Peters, D.M.; Conway, S.J.; Jefcoate, C.R.; Sheibani, N. Cyp1b1 Mediates Periostin Regulation of Trabecular Meshwork Development by Suppression of Oxidative Stress. Mol. Cell. Biol. 2013, 33, 4225–4240. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Han, Y.; Oatts, J.T. Genetic Changes and Testing Associated with Childhood Glaucoma: A Systematic Review. PLOS ONE 2024, 19, e0298883. [Google Scholar] [CrossRef] [PubMed]
- López-Garrido, M.-P.; Medina-Trillo, C.; Morales-Fernandez, L.; Garcia-Feijoo, J.; Martínez-de-la-Casa, J.-M.; García-Antón, M.; Escribano, J. Null CYP1B1 Genotypes in Primary Congenital and Nondominant Juvenile Glaucoma. Ophthalmology 2013, 120, 716–723. [Google Scholar] [CrossRef] [PubMed]
- Medina-Trillo, C.; Ferre-Fernández, J.-J.; Aroca-Aguilar, J.-D.; Bonet-Fernández, J.-M.; Escribano, J. Functional Characterization of Eight Rare Missense CYP1B1 Variants Involved in Congenital Glaucoma and Their Association with Null Genotypes. Acta Ophthalmol. (Copenh.) 2016, 94, e555–e560. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.O. Genetics of Primary Glaucoma. Curr. Opin. Ophthalmol. 2011, 22, 347–355. [Google Scholar] [CrossRef] [PubMed]
- Plásilová, M.; Stoilov, I.; Sarfarazi, M.; Kádasi, L.; Feráková, E.; Ferák, V. Identification of a Single Ancestral CYP1B1 Mutation in Slovak Gypsies (Roms) Affected with Primary Congenital Glaucoma. J. Med. Genet. 1999, 36, 290–294. [Google Scholar] [CrossRef] [PubMed]
- Kidson, S.H.; Kume, T.; Deng, K.; Winfrey, V.; Hogan, B.L. The Forkhead/Winged-Helix Gene, Mf1, Is Necessary for the Normal Development of the Cornea and Formation of the Anterior Chamber in the Mouse Eye. Dev. Biol. 1999, 211, 306–322. [Google Scholar] [CrossRef] [PubMed]
- Berry, F.B.; Lines, M.A.; Oas, J.M.; Footz, T.; Underhill, D.A.; Gage, P.J.; Walter, M.A. Functional Interactions between FOXC1 and PITX2 Underlie the Sensitivity to FOXC1 Gene Dose in Axenfeld-Rieger Syndrome and Anterior Segment Dysgenesis. Hum. Mol. Genet. 2006, 15, 905–919. [Google Scholar] [CrossRef] [PubMed]
- Paylakhi, S.H.; Moazzeni, H.; Yazdani, S.; Rassouli, P.; Arefian, E.; Jaberi, E.; Arash, E.H.; Gilani, A.S.; Fan, J.-B.; April, C.; et al. FOXC1 in Human Trabecular Meshwork Cells Is Involved in Regulatory Pathway That Includes miR-204, MEIS2, and ITGβ1. Exp. Eye Res. 2013, 111, 112–121. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Friedman, A.; Heaney, S.; Purcell, P.; Maas, R.L. Meis Homeoproteins Directly Regulate Pax6 during Vertebrate Lens Morphogenesis. Genes Dev. 2002, 16, 2097. [Google Scholar] [CrossRef] [PubMed]
- Medina-Trillo, C.; Sánchez-Sánchez, F.; Aroca-Aguilar, J.-D.; Ferre-Fernández, J.-J.; Morales, L.; Méndez-Hernández, C.-D.; Blanco-Kelly, F.; Ayuso, C.; García-Feijoo, J.; Escribano, J. Hypo- and Hypermorphic FOXC1 Mutations in Dominant Glaucoma: Transactivation and Phenotypic Variability. PLoS ONE 2015, 10, e0119272. [Google Scholar] [CrossRef] [PubMed]
- Harding, P.; Moosajee, M. The Molecular Basis of Human Anophthalmia and Microphthalmia. J. Dev. Biol. 2019, 7, 16. [Google Scholar] [CrossRef] [PubMed]
- Adler, R.; Canto-Soler, M.V. Molecular Mechanisms of Optic Vesicle Development: Complexities, Ambiguities and Controversies. Dev. Biol. 2007, 305, 1. [Google Scholar] [CrossRef] [PubMed]
- Gour, A.; Tibrewal, S.; Garg, A.; Vohra, M.; Ratna, R.; Sangwan, V.S. New Horizons in Aniridia Management: Clinical Insights and Therapeutic Advances. Taiwan J. Ophthalmol. 2023, 13, 467. [Google Scholar] [CrossRef] [PubMed]
- Schedl, A.; Ross, A.; Lee, M.; Engelkamp, D.; Rashbass, P.; van Heyningen, V.; Hastie, N.D. Influence of PAX6 Gene Dosage on Development: Overexpression Causes Severe Eye Abnormalities. Cell 1996, 86, 71–82. [Google Scholar] [CrossRef] [PubMed]
- Mort, R.L.; Bentley, A.J.; Martin, F.L.; Collinson, J.M.; Douvaras, P.; Hill, R.E.; Morley, S.D.; Fullwood, N.J.; West, J.D. Effects of Aberrant Pax6 Gene Dosage on Mouse Corneal Pathophysiology and Corneal Epithelial Homeostasis. PLoS ONE 2011, 6, e28895. [Google Scholar] [CrossRef] [PubMed]
- Gupta, V.; Somarajan, B.I.; Gupta, S.; Mahalingam, K.; Singh, A.; Sharma, A. A New Association of PAX6 Variation with Juvenile Onset Open Angle Glaucoma. J. Hum. Genet. 2023, 68, 355–358. [Google Scholar] [CrossRef] [PubMed]
- Haji-Seyed-Javadi, R.; Jelodari-Mamaghani, S.; Paylakhi, S.H.; Yazdani, S.; Nilforushan, N.; Fan, J.-B.; Klotzle, B.; Mahmoudi, M.J.; Ebrahimian, M.J.; Chelich, N.; et al. LTBP2 Mutations Cause Weill-Marchesani and Weill-Marchesani-like Syndrome and Affect Disruptions in the Extracellular Matrix. Hum. Mutat. 2012, 33, 1182–1187. [Google Scholar] [CrossRef] [PubMed]
- Narooie-Nejad, M.; Paylakhi, S.H.; Shojaee, S.; Fazlali, Z.; Rezaei Kanavi, M.; Nilforushan, N.; Yazdani, S.; Babrzadeh, F.; Suri, F.; Ronaghi, M.; et al. Loss of Function Mutations in the Gene Encoding Latent Transforming Growth Factor Beta Binding Protein 2, LTBP2, Cause Primary Congenital Glaucoma. Hum. Mol. Genet. 2009, 18, 3969–3977. [Google Scholar] [CrossRef] [PubMed]
- Gabriel, L.A.R.; Wang, L.W.; Bader, H.; Ho, J.C.; Majors, A.K.; Hollyfield, J.G.; Traboulsi, E.I.; Apte, S.S. ADAMTSL4, a Secreted Glycoprotein Widely Distributed in the Eye, Binds Fibrillin-1 Microfibrils and Accelerates Microfibril Biogenesis. Invest. Ophthalmol. Vis. Sci. 2012, 53, 461. [Google Scholar] [CrossRef] [PubMed]
- Cheong, S.-S.; Hentschel, L.; Davidson, A.E.; Gerrelli, D.; Davie, R.; Rizzo, R.; Pontikos, N.; Plagnol, V.; Moore, A.T.; Sowden, J.C.; et al. Mutations in CPAMD8 Cause a Unique Form of Autosomal-Recessive Anterior Segment Dysgenesis. Am. J. Hum. Genet. 2016, 99, 1338. [Google Scholar] [CrossRef] [PubMed]
- Escribano, J.; Tevar, A.; Bonet-Fernandez, J.-M.; Atienzar-Aroca, R.; Aroca-Aguilar, J.-D. Functional Interaction between Zebrafish Adamtsl4 and Cpamd8 Matrix Metalloproteinase-Related Genes: Implications in Ocular Anterior Segment Development and Glaucoma. Invest. Ophthalmol. Vis. Sci. 2024, 65, OD6. [Google Scholar]
- Abreu-Velez, A.M.; Howard, M.S. Collagen IV in Normal Skin and in Pathological Processes. North Am. J. Med. Sci. 2012, 4, 1. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.-W.; Kim, H.-K.; Naidansuren, P.; Ham, K.A.; Choi, H.S.; Ahn, H.-Y.; Kim, M.; Kang, D.H.; Kang, S.W.; Joe, Y.A. Peroxidasin Is Essential for Endothelial Cell Survival and Growth Signaling by Sulfilimine Crosslink-Dependent Matrix Assembly. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2020, 34, 10228–10241. [Google Scholar] [CrossRef] [PubMed]
- Bai, X.; Dilworth, D.J.; Weng, Y.-C.; Gould, D.B. Developmental Distribution of Collagen IV Isoforms and Relevance to Ocular Diseases. Matrix Biol. J. Int. Soc. Matrix Biol. 2009, 28, 194–201. [Google Scholar] [CrossRef] [PubMed]
- Kizhatil, K.; Ryan, M.; Marchant, J.K.; Henrich, S.; John, S.W.M. Schlemm’s Canal Is a Unique Vessel with a Combination of Blood Vascular and Lymphatic Phenotypes That Forms by a Novel Developmental Process. PLoS Biol. 2014, 12, e1001912. [Google Scholar] [CrossRef] [PubMed]
- Thomson, B.R.; Souma, T.; Tompson, S.W.; Onay, T.; Kizhatil, K.; Siggs, O.M.; Feng, L.; Whisenhunt, K.N.; Yanovitch, T.L.; Kalaydjieva, L.; et al. Angiopoietin-1 Is Required for Schlemm’s Canal Development in Mice and Humans. J. Clin. Invest. 2017, 127, 4421–4436. [Google Scholar] [CrossRef] [PubMed]
- Patel, S.P.; Parker, M.D. SLC4A11 and the Pathophysiology of Congenital Hereditary Endothelial Dystrophy. BioMed Res. Int. 2015, 2015, 475392. [Google Scholar] [CrossRef] [PubMed]
- Vilas, G.L.; Loganathan, S.K.; Liu, J.; Riau, A.K.; Young, J.D.; Mehta, J.S.; Vithana, E.N.; Casey, J.R. Transmembrane Water-Flux through SLC4A11: A Route Defective in Genetic Corneal Diseases. Hum. Mol. Genet. 2013, 22, 4579–4590. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Sun, M.; Zhang, Q.; Dang, S.; Zhang, W. ADAMTS18 Regulates Early Branching Morphogenesis of Lacrimal Gland and Has a Significant Association with the Risk of Dry Eye in Mice. Exp. Eye Res. 2022, 218, 109020. [Google Scholar] [CrossRef] [PubMed]
- Ataca, D.; Caikovski, M.; Piersigilli, A.; Moulin, A.; Benarafa, C.; Earp, S.E.; Guri, Y.; Kostic, C.; Arsenijevic, Y.; Soininen, R.; et al. Adamts18 Deletion Results in Distinct Developmental Defects and Provides a Model for Congenital Disorders of Lens, Lung, and Female Reproductive Tract Development. Biol. Open 2016, 5, 1585–1594. [Google Scholar] [CrossRef] [PubMed]
- Aldahmesh, M.A.; Alshammari, M.J.; Khan, A.O.; Mohamed, J.Y.; Alhabib, F.A.; Alkuraya, F.S. The Syndrome of Microcornea, Myopic Chorioretinal Atrophy, and Telecanthus (MMCAT) Is Caused by Mutations in ADAMTS18. Hum. Mutat. 2013, 34, 1195–1199. [Google Scholar] [CrossRef] [PubMed]
- Aldahmesh, M.A.; Khan, A.O.; Mohamed, J.Y.; Alkuraya, H.; Ahmed, H.; Bobis, S.; Al-Mesfer, S.; Alkuraya, F.S. Identification of ADAMTS18 as a Gene Mutated in Knobloch Syndrome. J. Med. Genet. 2011, 48, 597–601. [Google Scholar] [CrossRef] [PubMed]
- Cvekl, A.; Camerino, M.J. Generation of Lens Progenitor Cells and Lentoid Bodies from Pluripotent Stem Cells: Novel Tools for Human Lens Development and Ocular Disease Etiology. Cells 2022, 11, 3516. [Google Scholar] [CrossRef] [PubMed]
- Semina, E.V.; Ferrell, R.E.; Mintz-Hittner, H.A.; Bitoun, P.; Alward, W.L.M.; Reiter, R.S.; Funkhauser, C.; Daack-Hirsch, S.; Murray, J.C. A Novel Homeobox Gene PITX3 Is Mutated in Families with Autosomal-Dominant Cataracts and ASMD. Nat. Genet. 1998, 19, 167–170. [Google Scholar] [CrossRef] [PubMed]
- Dodonova, S.O.; Zhu, F.; Dienemann, C.; Taipale, J.; Cramer, P. Nucleosome-Bound SOX2 and SOX11 Structures Elucidate Pioneer Factor Function. Nature 2020, 580, 669–672. [Google Scholar] [CrossRef] [PubMed]
- Cizelsky, W.; Hempel, A.; Metzig, M.; Tao, S.; Hollemann, T.; Kühl, M.; Kühl, S.J. Sox4 And Sox11 Function during Xenopus Laevis Eye Development. PLOS ONE 2013, 8, e69372. [Google Scholar] [CrossRef] [PubMed]
- Tamm, E.R.; Wurm, A.; Sock, E.; Fuchshofer, R.; Wegner, M. The High–Mobility–Group Transcription Factor Sox11 Plays an Important Role During Anterior Eye Segment Development. Invest. Ophthalmol. Vis. Sci. 2006, 47, 3874. [Google Scholar]
- Li, L.; Fan, D.-B.; Zhao, Y.-T.; Li, Y.; Yang, Z.-B.; Zheng, G.-Y. GJA8 Missense Mutation Disrupts Hemichannels and Induces Cell Apoptosis in Human Lens Epithelial Cells. Sci. Rep. 2019, 9, 19157. [Google Scholar] [CrossRef] [PubMed]
- Kao, W.W.-Y.; Liu, C.-Y. Roles of Lumican and Keratocan on Corneal Transparency. Glycoconj. J. 2002, 19, 275–285. [Google Scholar] [CrossRef] [PubMed]
- Conrad, A.H.; Conrad, G.W. The Keratocan Gene Is Expressed in Both Ocular and Non-Ocular Tissues during Early Chick Development. Matrix Biol. 2003, 22, 323–337. [Google Scholar] [CrossRef] [PubMed]
- Gealy, E.C.; Kerr, B.C.; Young, R.D.; Tudor, D.; Hayes, A.J.; Hughes, C.E.; Caterson, B.; Quantock, A.J.; Ralphs, J.R. Differential Expression of the Keratan Sulphate Proteoglycan, Keratocan, during Chick Corneal Embryogenesis. Histochem. Cell Biol. 2007, 128, 551–555. [Google Scholar] [CrossRef] [PubMed]
- Pellegata, N.S.; Dieguez-Lucena, J.L.; Joensuu, T.; Lau, S.; Montgomery, K.T.; Krahe, R.; Kivelä, T.; Kucherlapati, R.; Forsius, H.; de la Chapelle, A. Mutations in KERA, Encoding Keratocan, Cause Cornea Plana. Nat. Genet. 2000, 25, 91–95. [Google Scholar] [CrossRef] [PubMed]
- Accogli, A.; Calabretta, S.; St-Onge, J.; Boudrahem-Addour, N.; Dionne-Laporte, A.; Joset, P.; Azzarello-Burri, S.; Rauch, A.; Krier, J.; Fieg, E.; et al. De Novo Pathogenic Variants in N-Cadherin Cause a Syndromic Neurodevelopmental Disorder with Corpus Callosum, Axon, Cardiac, Ocular, and Genital Defects. Am. J. Hum. Genet. 2019, 105, 854. [Google Scholar] [CrossRef] [PubMed]
- Karwacki-Neisius, V.; Jang, A.; Cukuroglu, E.; Tai, A.; Jiao, A.; Predes, D.; Yoon, J.; Brookes, E.; Chen, J.; Iberg, A.; et al. WNT Signalling Control by KDM5C during Development Affects Cognition. Nature 2024, 627, 594–603. [Google Scholar] [CrossRef] [PubMed]
- Schorle, H.; Meier, P.; Buchert, M.; Jaenisch, R.; Mitchell, P.J. Transcription Factor AP-2 Essential for Cranial Closure and Craniofacial Development. Nature 1996, 381, 235–238. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Jeong, Y.; Kwon, K.; Ismail, T.; Lee, H.-K.; Kim, C.; Park, J.-W.; Kwon, O.-S.; Kang, B.-S.; Lee, D.-S.; et al. Physiological Effects of KDM5C on Neural Crest Migration and Eye Formation during Vertebrate Development. Epigenetics Chromatin 2018, 11, 72. [Google Scholar] [CrossRef] [PubMed]
- Gestri, G.; Osborne, R.J.; Wyatt, A.W.; Gerrelli, D.; Gribble, S.; Stewart, H.; Fryer, A.; Bunyan, D.J.; Prescott, K.; Collin, J.R.O.; et al. Reduced TFAP2A Function Causes Variable Optic Fissure Closure and Retinal Defects and Sensitizes Eye Development to Mutations in Other Morphogenetic Regulators. Hum. Genet. 2009, 126, 791–803. [Google Scholar] [CrossRef] [PubMed]
- Shao, F.; Phan, A.-V.; Yu, W.; Guo, Y.; Thompson, J.; Coppinger, C.; Venugopalan, S.R.; Amendt, B.A.; Van Otterloo, E.; Cao, H. Transcriptional Programs of Pitx2 and Tfap2a/Tfap2b Controlling Lineage Specification of Mandibular Epithelium during Tooth Initiation. PLOS Genet. 2024, 20, e1011364. [Google Scholar] [CrossRef] [PubMed]
- Mandal, A.K.; Chakrabarti, D.; Gothwal, V.K. Approach to Primary Congenital Glaucoma: A Perspective. Taiwan J. Ophthalmol. 2023, 13, 451. [Google Scholar] [CrossRef] [PubMed]
- Coviltir, V.; Marinescu, M.C.; Urse, B.M.; Burcel, M.G. Primary Congenital and Childhood Glaucoma—A Complex Clinical Picture and Surgical Management. Diagnostics 2025, 15, 308. [Google Scholar] [CrossRef] [PubMed]
- Hashemi, H.; Ghaffari, R.; Mohebi, M. Posterior Lamellar Keratoplasty (DSAEK) in Peters Anomaly. Cornea 2012, 31, 1201–1205. [Google Scholar] [CrossRef] [PubMed]
- Research, C. for B.E. and Approved Cellular and Gene Therapy Products. FDA 2024. [Google Scholar]
- Amador, C.; Shah, R.; Ghiam, S.; Kramerov, A.A.; Ljubimov, A.V. Gene Therapy in the Anterior Eye Segment. Curr. Gene Ther. 2022, 22, 104–131. [Google Scholar] [CrossRef] [PubMed]
- Mirjalili Mohanna, S.Z.; Korecki, A.J.; Simpson, E.M. rAAV-PHP.B Escapes the Mouse Eye and Causes Lethality Whereas rAAV9 Can Transduce Aniridic Corneal Limbal Stem Cells without Lethality. Gene Ther. 2023, 30, 670–684. [Google Scholar] [CrossRef] [PubMed]
- Jain, A.; Zode, G.; Kasetti, R.B.; Ran, F.A.; Yan, W.; Sharma, T.P.; Bugge, K.; Searby, C.C.; Fingert, J.H.; Zhang, F.; et al. CRISPR-Cas9–Based Treatment of Myocilin-Associated Glaucoma. Proc. Natl. Acad. Sci. USA 2017, 114, 11199–11204. [Google Scholar] [CrossRef] [PubMed]
- Behrens, A.; Gordon, E.M.; Li, L.; Liu, P.X.; Chen, Z.; Peng, H.; La Bree, L.; Anderson, W.F.; Hall, F.L.; McDonnell, P.J. Retroviral Gene Therapy Vectors for Prevention of Excimer Laser-Induced Corneal Haze. Invest. Ophthalmol. Vis. Sci. 2002, 43, 968–977. [Google Scholar] [PubMed]
- Patil, S.V.; Kaipa, B.R.; Ranshing, S.; Sundaresan, Y.; Millar, J.C.; Nagarajan, B.; Kiehlbauch, C.; Zhang, Q.; Jain, A.; Searby, C.C.; et al. Lentiviral Mediated Delivery of CRISPR/Cas9 Reduces Intraocular Pressure in a Mouse Model of Myocilin Glaucoma. Sci. Rep. 2024, 14, 6958. [Google Scholar] [CrossRef] [PubMed]
- Tandon, A.; Sharma, A.; Rodier, J.T.; Klibanov, A.M.; Rieger, F.G.; Mohan, R.R. BMP7 Gene Transfer via Gold Nanoparticles into Stroma Inhibits Corneal Fibrosis In Vivo. PLoS ONE 2013, 8, e66434. [Google Scholar] [CrossRef] [PubMed]
- Tong, Y.-C.; Chang, S.-F.; Liu, C.-Y.; Kao, W.W.-Y.; Huang, C.H.; Liaw, J. Eye Drop Delivery of Nano-Polymeric Micelle Formulated Genes with Cornea-Specific Promoters. J. Gene Med. 2007, 9, 956–966. [Google Scholar] [CrossRef] [PubMed]
- Yoon, K.C.; Bae, J.A.; Park, H.J.; Im, S.K.; Oh, H.J.; Lin, X.H.; Kim, M.Y.; Lee, J.H.; Lee, S.E.; Ahn, K.Y.; et al. Subconjunctival Gene Delivery of the Transcription Factor GA-Binding Protein Delays Corneal Neovascularization in a Mouse Model. Gene Ther. 2009, 16, 973–981. [Google Scholar] [CrossRef] [PubMed]
- Blair-Parks, K.; Weston, B.C.; Dean, D.A. High-Level Gene Transfer to the Cornea Using Electroporation. J. Gene Med. 2002, 4, 92–100. [Google Scholar] [CrossRef] [PubMed]
- Vinciguerra, P.; Romano, V.; Rosetta, P.; Legrottaglie, E.F.; Kubrak-Kisza, M.; Azzolini, C.; Vinciguerra, R. Iontophoresis-Assisted Corneal Collagen Cross-Linking with Epithelial Debridement: Preliminary Results. BioMed Res. Int. 2016, 2016, 3720517. [Google Scholar] [CrossRef] [PubMed]
- Henderson, J.; O’Callaghan, J.; Campbell, M. Gene Therapy for Glaucoma: Targeting Key Mechanisms. Vision Res. 2024, 225, None. [Google Scholar] [CrossRef] [PubMed]
- Sulak, R.; Liu, X.; Smedowski, A. The Concept of Gene Therapy for Glaucoma: The Dream That Has Not Come True Yet. Neural Regen. Res. 2023, 19, 92–99. [Google Scholar] [CrossRef] [PubMed]
- O’Callaghan, J.; Delaney, C.; O’Connor, M.; van Batenburg-Sherwood, J.; Schicht, M.; Lütjen-Drecoll, E.; Hudson, N.; Ni Dhubhghaill, S.; Humphries, P.; Stanley, C.; et al. Matrix Metalloproteinase-3 (MMP-3)–Mediated Gene Therapy for Glaucoma. Sci. Adv. 2023, 9, eadf6537. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Bell, O.H.; Copland, D.A.; Young, A.; Pooley, J.R.; Maswood, R.; Evans, R.S.; Khaw, P.T.; Ali, R.R.; Dick, A.D.; et al. Gene Therapy for Glaucoma by Ciliary Body Aquaporin 1 Disruption Using CRISPR-Cas9. Mol. Ther. 2020, 28, 820. [Google Scholar] [CrossRef] [PubMed]
- Tan, C.; Song, M.; Stamer, W.D.; Qiao, Y.; Chen, X.; Sun, X.; Lei, Y.; Chen, J. miR-21-5p: A Viable Therapeutic Strategy for Regulating Intraocular Pressure. Exp. Eye Res. 2020, 200, 108197. [Google Scholar] [CrossRef] [PubMed]
- Wójcik-Gryciuk, A.; Gajewska-Woźniak, O.; Kordecka, K.; Boguszewski, P.M.; Waleszczyk, W.; Skup, M. Neuroprotection of Retinal Ganglion Cells with AAV2-BDNF Pretreatment Restoring Normal TrkB Receptor Protein Levels in Glaucoma. Int. J. Mol. Sci. 2020, 21, 6262. [Google Scholar] [CrossRef] [PubMed]
- Jiang, W.; Tang, L.; Zeng, J.; Chen, B. Adeno-Associated Virus Mediated SOD Gene Therapy Protects the Retinal Ganglion Cells from Chronic Intraocular Pressure Elevation Induced Injury via Attenuating Oxidative Stress and Improving Mitochondrial Dysfunction in a Rat Model. Am. J. Transl. Res. 2016, 8, 799–810. [Google Scholar] [PubMed]
- Richardson, R.; Smart, M.; Tracey-White, D.; Webster, A.R.; Moosajee, M. Mechanism and Evidence of Nonsense Suppression Therapy for Genetic Eye Disorders. Exp. Eye Res. 2017, 155, 24–37. [Google Scholar] [CrossRef] [PubMed]
- “pax6” [GENE] - ClinVar - NCBI. Available online: https://www.ncbi.nlm.nih.gov/clinvar (accessed on 27 January 2025).
- Lima Cunha, D.; Sarkar, H.; Eintracht, J.; Harding, P.; Zhou, J.H.; Moosajee, M. Restoration of Functional PAX6 in Aniridia Patient iPSC-Derived Ocular Tissue Models Using Repurposed Nonsense Suppression Drugs. Mol. Ther. Nucleic Acids 2023, 33, 240–253. [Google Scholar] [CrossRef] [PubMed]
- Gregory-Evans, C.Y.; Wang, X.; Wasan, K.M.; Zhao, J.; Metcalfe, A.L.; Gregory-Evans, K. Postnatal Manipulation of Pax6 Dosage Reverses Congenital Tissue Malformation Defects. J. Clin. Invest. 2014, 124, 111–116. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Gregory-Evans, K.; Wasan, K.M.; Sivak, O.; Shan, X.; Gregory-Evans, C.Y. Efficacy of Postnatal In Vivo Nonsense Suppression Therapy in a Pax6 Mouse Model of Aniridia. Mol. Ther. Nucleic Acids 2017, 7, 417–428. [Google Scholar] [CrossRef] [PubMed]
- PTC Therapeutics. A Phase 2, Multicenter, Randomized, Double-Masked, Placebo-Controlled Study of the Safety and Efficacy of Ataluren (PTC124) for the Treatment of Nonsense Mutation Aniridia; clinicaltrials.gov; 2022.
- Mirjalili Mohanna, S.Z.; Hickmott, J.W.; Lam, S.L.; Chiu, N.Y.; Lengyell, T.C.; Tam, B.M.; Moritz, O.L.; Simpson, E.M. Germline CRISPR/Cas9-Mediated Gene Editing Prevents Vision Loss in a Novel Mouse Model of Aniridia. Mol. Ther. Methods Clin. Dev. 2020, 17, 478–490. [Google Scholar] [CrossRef] [PubMed]
- Korecki, A.J.; Cueva-Vargas, J.L.; Fornes, O.; Agostinone, J.; Farkas, R.A.; Hickmott, J.W.; Lam, S.L.; Mathelier, A.; Zhou, M.; Wasserman, W.W.; et al. Human MiniPromoters for Ocular-rAAV Expression in ON Bipolar, Cone, Corneal, Endothelial, Müller Glial, and PAX6 Cells. Gene Ther. 2021, 28, 351–372. [Google Scholar] [CrossRef] [PubMed]
- Dorot, O.; Roux, L.N.; Zennaro, L.; Oved, K.; Bremond-Gignac, D.; Pichinuk, E.; Aberdam, D. The Antipsychotropic Drug Duloxetine Rescues PAX6 Haploinsufficiency of Mutant Limbal Stem Cells through Inhibition of the MEK/ERK Signaling Pathway. Ocul. Surf. 2022, 23, 140–142. [Google Scholar] [CrossRef] [PubMed]
- Oved, K.; Zennaro, L.; Dorot, O.; Zerbib, J.; Frank, E.; Roux, L.N.; Bremond-Gignac, D.; Pichinuk, E.; Aberdam, D. Ritanserin, a Potent Serotonin 2A Receptor Antagonist, Represses MEK/ERK Signalling Pathway to Restore PAX6 Production and Function in Aniridia-like Cellular Model. Biochem. Biophys. Res. Commun. 2021, 582, 100–104. [Google Scholar] [CrossRef] [PubMed]
- Moustardas, P.; Abbasi, M.; Javidjam, D.; Asamoah, C.S.; Schweitzer-Chaput, A.; Cisternino, S.; Bremond-Gignac, D.; Aberdam, D.; Lagali, N. Duloxetine Enhances PAX6 Expression and Suppresses Innate Immune Responses in Murine LPS-Induced Corneal Inflammation. Ocul. Surf. 2024, 34, 225–234. [Google Scholar] [CrossRef] [PubMed]
- Abbasi, M.; Amini, M.; Moustardas, P.; Gutsmiedl, Q.; Javidjam, D.; Suiwal, S.; Seitz, B.; Fries, F.N.; Dashti, A.; Rautavaara, Y.; et al. Effects of miR-204-5p Modulation on PAX6 Regulation and Corneal Inflammation. Sci. Rep. 2024, 14, 26436. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Anterior segment embryological development: (a) At day 37 of gestation a double layered optic cup and lens vesicle can be seen; (b) At 47 days the optic cup has formed primitive neural retina and retinal pigment epithelium with the optic stalk forming primitive optic nerve. Optic cup patterning is dependent on the PAX6 expression gradient, from high expression at the peripheral optic cup tips down to low expression centrally. Cells of the central lens placode migrate to the posterior lens vesicle and elongate to form primary lens fibre cells, and the first wave of neural crest cells from the periocular mesenchyme migrates between the lens and surface ectoderm; (c) At 57 days the primitive corneal epithelium is formed from the surface ectoderm, the corneal stroma and endothelium are formed from neural crest cells of the periocular mesenchyme, there is separation of the lens stalk and formation of the anterior chamber; (d) By 15 weeks elongation of optic cup tips along the anterior lens surface to form the iris and ciliary body epithelium occurs, and there is a second wave of periocular mesenchyme migration into the anterior chamber to form the iris and ciliary body stroma; (e) At 20 weeks there is an accumulation of periocular mesenchyme in the iridocorneal angle which subsequently elongates and forms lamellae to become the trabecular meshwork, and vessels form in the adjacent sclera which will become Schlemm’s canal. Gene expression marked in red. RPE, retinal pigment epithelium.
Figure 1.
Anterior segment embryological development: (a) At day 37 of gestation a double layered optic cup and lens vesicle can be seen; (b) At 47 days the optic cup has formed primitive neural retina and retinal pigment epithelium with the optic stalk forming primitive optic nerve. Optic cup patterning is dependent on the PAX6 expression gradient, from high expression at the peripheral optic cup tips down to low expression centrally. Cells of the central lens placode migrate to the posterior lens vesicle and elongate to form primary lens fibre cells, and the first wave of neural crest cells from the periocular mesenchyme migrates between the lens and surface ectoderm; (c) At 57 days the primitive corneal epithelium is formed from the surface ectoderm, the corneal stroma and endothelium are formed from neural crest cells of the periocular mesenchyme, there is separation of the lens stalk and formation of the anterior chamber; (d) By 15 weeks elongation of optic cup tips along the anterior lens surface to form the iris and ciliary body epithelium occurs, and there is a second wave of periocular mesenchyme migration into the anterior chamber to form the iris and ciliary body stroma; (e) At 20 weeks there is an accumulation of periocular mesenchyme in the iridocorneal angle which subsequently elongates and forms lamellae to become the trabecular meshwork, and vessels form in the adjacent sclera which will become Schlemm’s canal. Gene expression marked in red. RPE, retinal pigment epithelium.
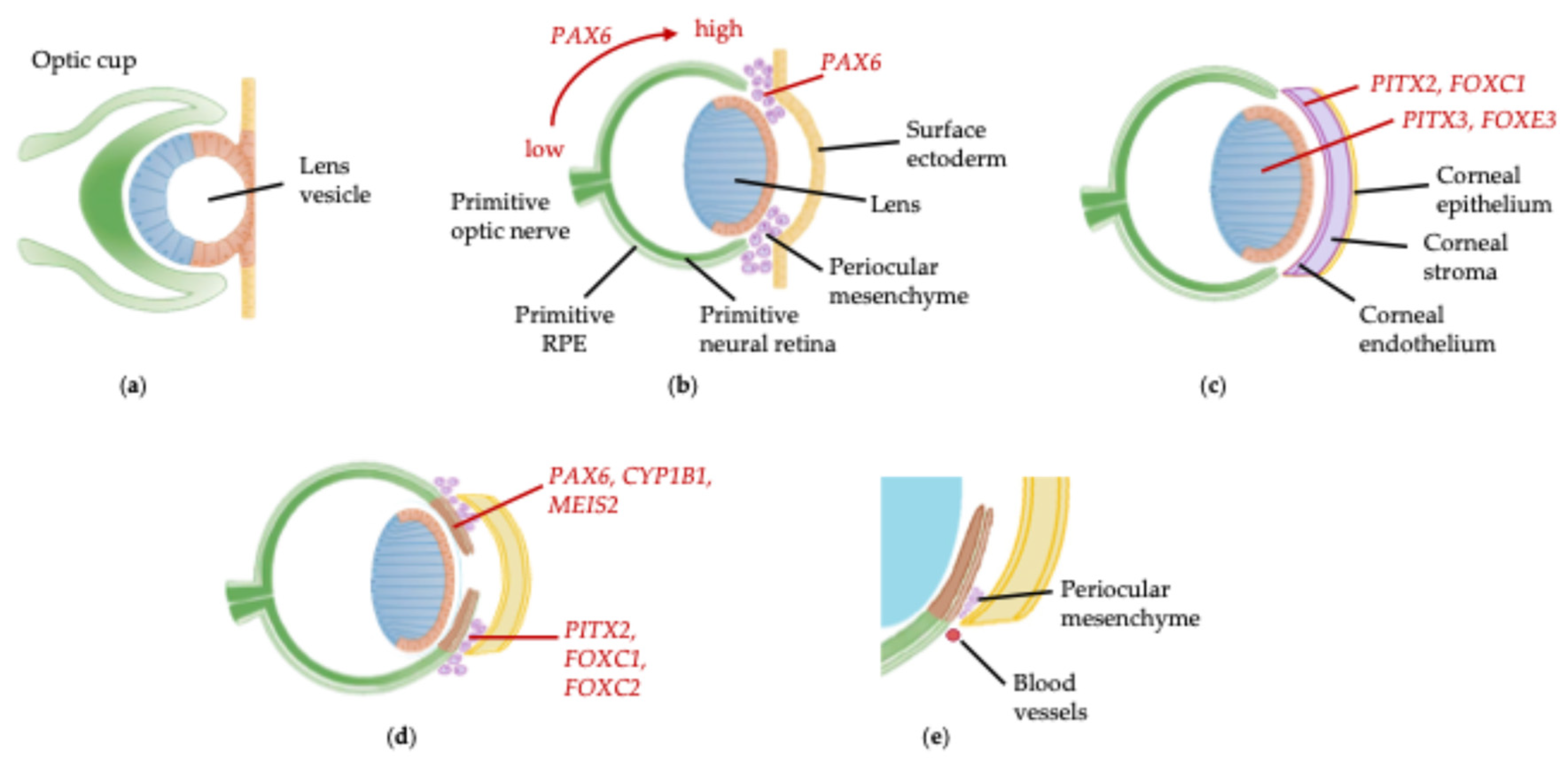
Figure 2.
Clinical features of childhood glaucoma and anterior segment dysgenesis: (a) Right buphthalmos due to unilateral primary congenital glaucoma (PCG); (b) Immature scalloped appearance of iridocorneal angle in PCG; (c) Central corneal opacity in Peters anomaly; (d-e) Axenfeld-Rieger anomaly: (d) iris hypoplasia and posterior embryotoxon (white arrowhead); (e) Corectopia; (f-h) Congenital aniridia: (f) iris hypoplasia; (g) foveal hypoplasia; (h) aniridia-associated keratopathy.
Figure 2.
Clinical features of childhood glaucoma and anterior segment dysgenesis: (a) Right buphthalmos due to unilateral primary congenital glaucoma (PCG); (b) Immature scalloped appearance of iridocorneal angle in PCG; (c) Central corneal opacity in Peters anomaly; (d-e) Axenfeld-Rieger anomaly: (d) iris hypoplasia and posterior embryotoxon (white arrowhead); (e) Corectopia; (f-h) Congenital aniridia: (f) iris hypoplasia; (g) foveal hypoplasia; (h) aniridia-associated keratopathy.
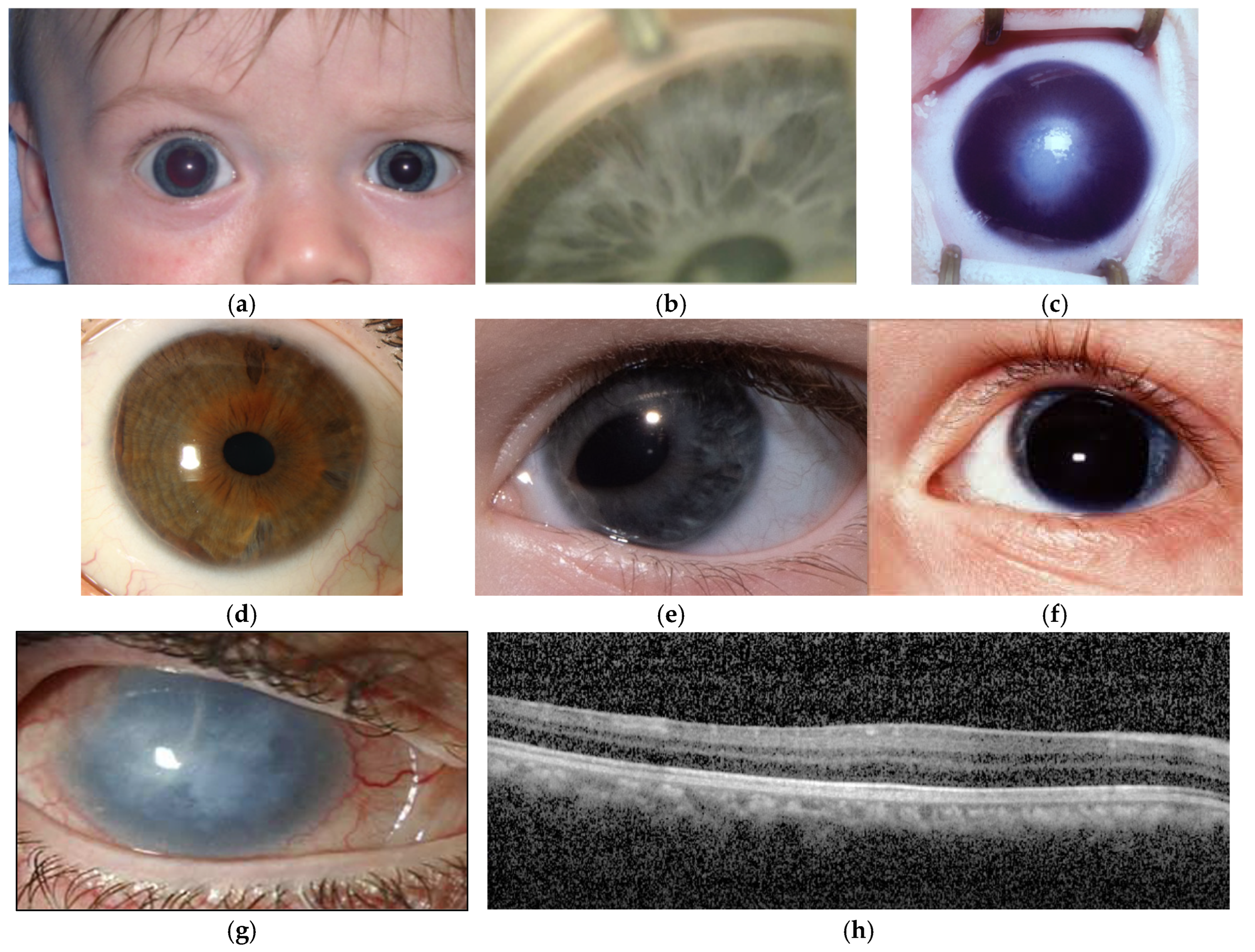
Figure 3.
Ocular expression of genes causing non-syndromic childhood glaucoma and anterior segment dysgenesis.
Figure 3.
Ocular expression of genes causing non-syndromic childhood glaucoma and anterior segment dysgenesis.
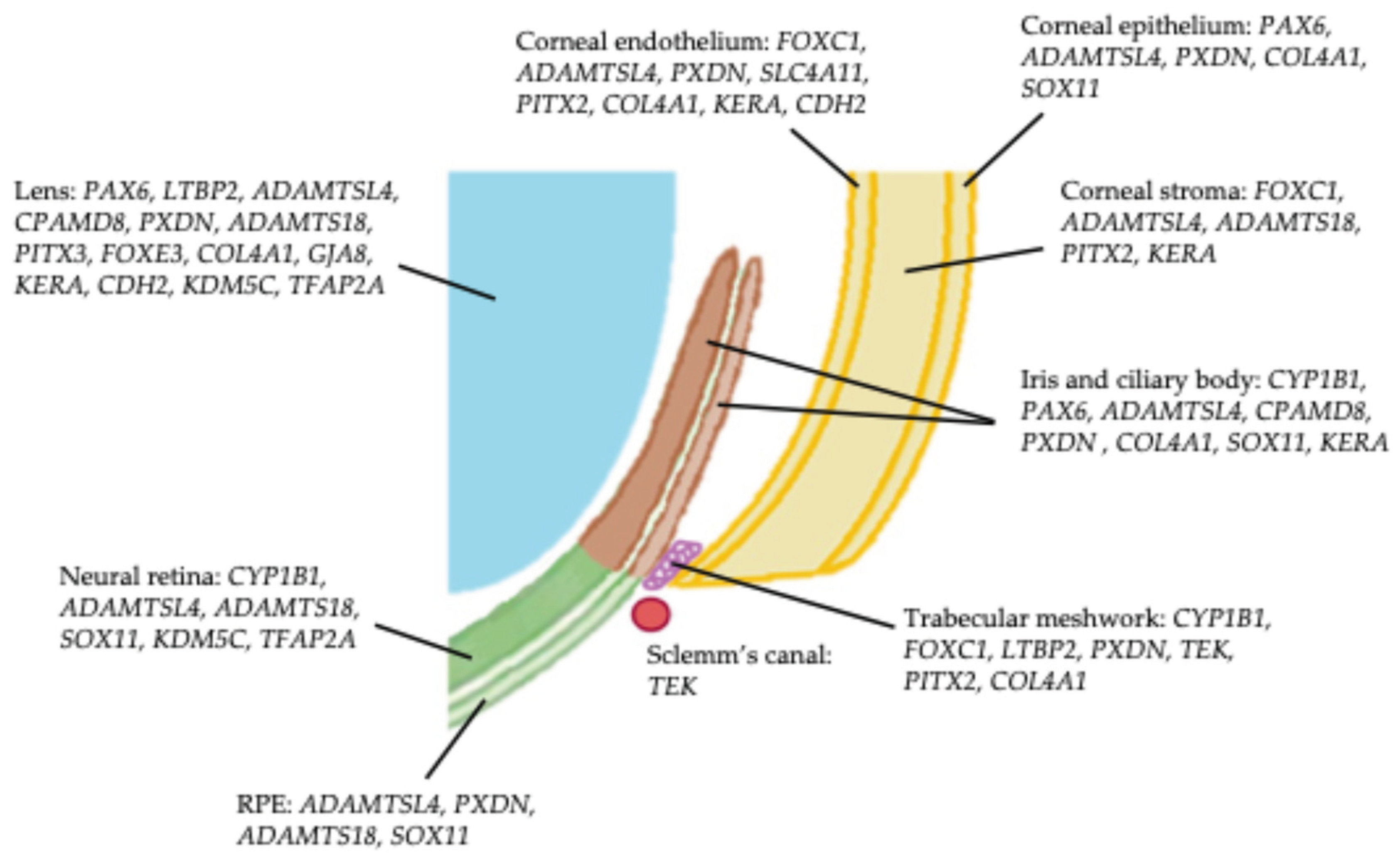

Table 1.
Genes reported to be linked to childhood glaucoma associated with anterior segment dysgenesis, and the phenotypes identified in each.
Table 1.
Genes reported to be linked to childhood glaucoma associated with anterior segment dysgenesis, and the phenotypes identified in each.
| Gene | OMIM ID | Phenotype(s) of anterior segment dysgenesis associated with childhood glaucoma | Associated with isolated childhood glaucoma |
|---|---|---|---|
| CYP1B1 | 601771 | Peters anomaly [20,21,22,23], Axenfeld-Rieger anomaly [24], aniridia [25], corneal dystrophy [12,26], unclassified ASD [27]. | Yes |
| FOXC1 | 601090 | Peters anomaly [13,28], Axenfeld-Rieger anomaly [15,23,29], aniridia [13], unclassified ASD [12,13]. | Yes |
| PAX6 | 607108 | Aniridia [16,29,30], Peters anomaly [28]. | Yes |
| LTBP2 | 602091 | Weill-Marchesani syndrome [23,31], congenital cataract [12], lenticular anomalies [16,32], unclassified ASD [16]. | Yes |
| ADAMTSL4 | 610113 | Ectopia lentis [33]. | Yes |
| CPAMD8 | 608841 | Lenticular anomalies [34,35], iris anomalies [35], unclassified ASD [16,36]. | Yes |
| PXDN | 605158 | Peters anomaly [37], congenital cataract [38], aphakia [37], aniridia [39], sclerocornea [28,40]. | Yes |
| TEK | 600221 | Sclerocornea [31]. | Yes |
| SLC4A11 | 610206 | Congenital hereditary endothelial dystrophy [16,27,41]. | Yes |
| ADAMTS18 | 607512 | Microcornea [42]. | Yes |
| PITX3 | 602669 | Microcornea [43,44], congenital cataract [44]. | Yes |
| PITX2 | 601542 | Peters anomaly [13,16], Axenfeld-Rieger anomaly [16,28], sclerocornea [28]. | No |
| FOXE3 | 601094 | Peters anomaly [45,46,47], iris anomalies [45,46], congenital cataract/aphakia [46,48], microphthalmia [46,49]. | No |
| COL4A1 | 120130 | Congenital cataract [13,50,51], Axenfeld-Rieger anomaly [51], iris anomalies [13,50]. | No |
| SOX11 | 600898 | Peters anomaly [52], aniridia [31,52]. | No |
| GJA8 | 600897 | Sclerocornea [23,28,31]. | No |
| KERA | 603288 | Unclassified ASD [34]. | No |
| CDH2 | 114020 | Peters anomaly [53]. | No |
| KDM5C | 314690 | Peters anomaly [54]. | No |
| TFAP2A | 107580 | Peters anomaly [55]. | No |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



