Submitted:
18 July 2025
Posted:
18 July 2025
You are already at the latest version
Abstract
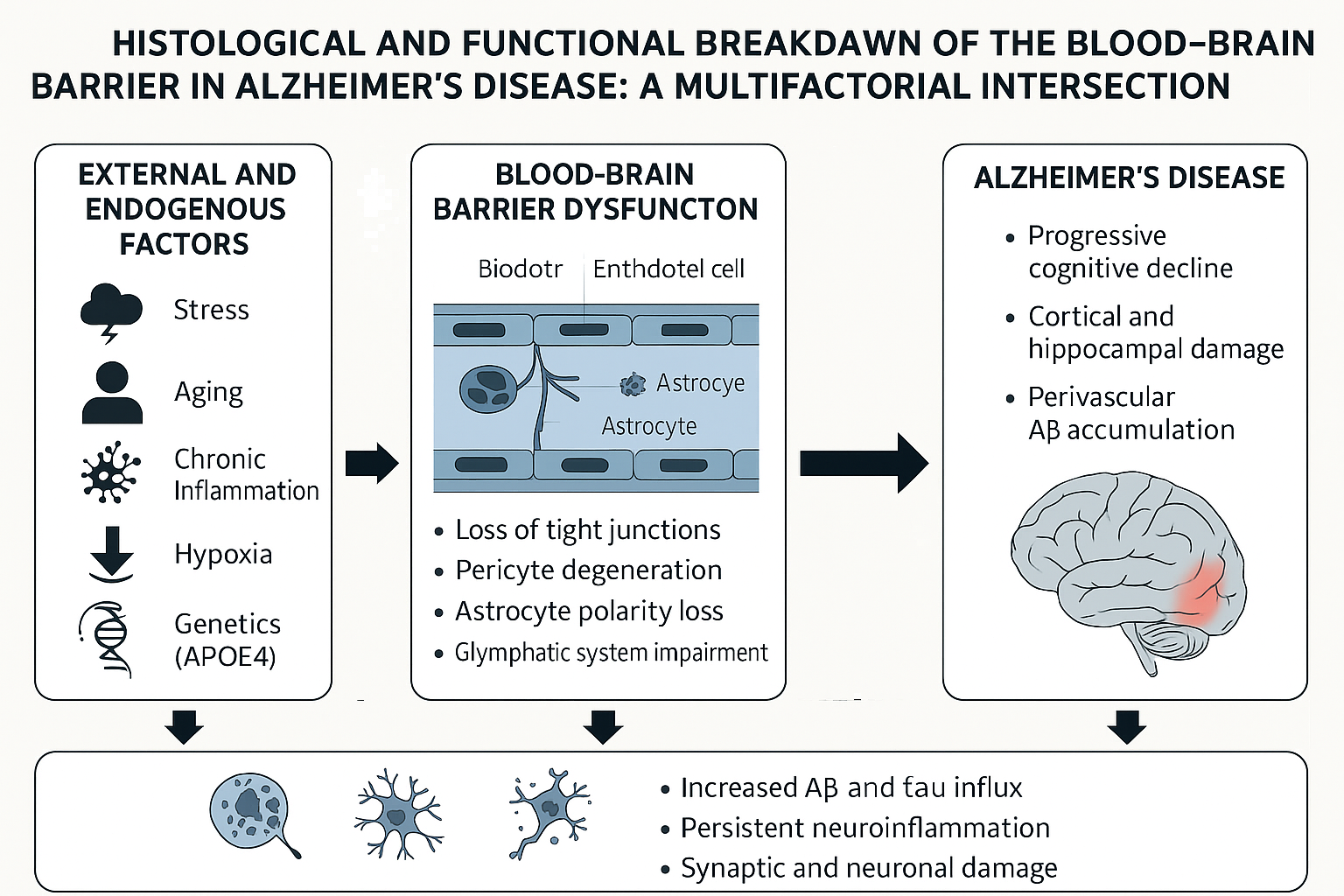
Background: Alzheimer’s disease (AD) is a multifactorial neurodegenerative disorder characterized by beta-amyloid plaques, neurofibrillary tangles, and progressive cognitive decline. Recent evidence has highlighted the role of blood–brain barrier (BBB) dysfunction in the early stages of AD pathology. Objective: To explore the histological structure and physiological function of the blood–brain barrier, and to identify the shared pathological mechanisms between BBB disruption and Alzheimer’s disease progression. Methods: This narrative review was conducted through a comprehensive search of peer-reviewed literature from 1997 to 2024, using databases such as PubMedh, Elsevier, Scopus, and Google Scholar. Results: Multiple histological and cellular components—including endothelial cells, pericytes, astrocytes, and tight junctions—contribute to BBB integrity. The breakdown of this barrier in AD is associated with chronic inflammation, oxidative stress, vascular injury, pericyte degeneration, astrocyte polarity loss, and dysfunction of nutrient transport systems like GLUT1. These alterations promote neuroinflammation, beta-amyloid accumulation, and progressive neuronal damage. Conclusion: BBB dysfunction is not merely a consequence of AD but may act as an early and active driver of its pathogenesis. Understanding the mechanisms of BBB breakdown can lead to early diagnostic markers and novel therapeutic strategies aimed at preserving or restoring barrier integrity in Alzheimer’s disease.
Keywords:
blood-brain barrier
; Alzheimer's disease
; degeneration
; histological
; microglia
; inflammation
; vascular damage
; oxidative stress
Introduction
All higher beings constantly defend their biological integrity against a series of essentially external aggression. Thus, cells and molecules of the immune system reach most tissues through the bloodstream, penetrating through the walls of blood capillaries [1]. Similarly, the nervous system also penetrates most tissues, but the two systems avoid each other because the blood-brain barrier prevents lymphocytes (the most well-known immune cells) from coming into contact with neurons [2,3,4,5]. This is because lymphocytes can secrete substances capable of destroying neurons, which is why many researchers believe that the nervous system lacks immune protection. However, the importance of the nervous system in proper functioning is well known. In fact, a mechanism is needed to protect the system that gives us life from infections and neurodegenerative diseases, including Alzheimer's disease [1,2,4].
About 30 million people in the world suffer from Alzheimer's neurodegenerative disease, according to the World Health Organization (WHO) [10].
A weakening of the BBB could allow greater infiltration of beta-amyloid in the brain, thus complementing that produced in the brain itself and accelerating neuronal deterioration, which is related to the onset of Alzheimer's disease [9].
Method
This narrative review focused on the relationship between blood-brain barrier (BBB) dysfunction and Alzheimer's disease (AD). Information was collected from scientific literature published between 1997 and 2024, including articles in peer-reviewed scientific journals, textbooks, and reports from prestigious health organizations. The PubMed, Elsevier, Scopus, and Google Scholar databases were used for the literature search. The search terms included "blood-brain barrier," "Alzheimer's disease," "BBB permeability," "neuroinflammation," and "vascular dysfunction." vascular dysfunction. Articles were selected based on their relevance to the histological structure of the BBB, mechanisms of BBB degradation, and relationship to Alzheimer's pathology. Studies focusing on human or mammalian models, addressing BBB damage mechanisms or analyzing the contribution of the BBB were included.
Historical Data
Blood-Brain Barrier
The first indication of the existence of the BBB was reported by Paul Ehrlich in 1885. Ehrlich observed that the injection of a dye (e.g., methylene blue) into the venous or arterial vascular network of rabbits stained all tissues in the animal's body, except the nervous tissue. Shortly thereafter, one of Ehrlich's collaborators, Edwin E. Goldmann, stained the brain but not the body tissues [7]. This finding has led to studies to determine the anatomical, biochemical, or physiological factors involved in this separation between brain tissue, cerebrospinal fluid, and blood circulation. It was not until 1967, thanks to electron microscopy studies by Karnovsky et al., who found that the capillary endothelium of the brain had tightly connected intercellular junctions, and that the morphological structure that constitutes the BBB was located there. [8,9]
Alzheimer's
In 1901, German psychiatrist Alois Alzheimer discovered the first case of what is now known as Alzheimer's disease in a 51-year-old woman. The researcher followed his patient until her death in 1906 and was then able to observe the brain [5]. After this time, the patient was able to report the case publicly for the first time. After the death, her brain was examined under a microscope. He observed changes in the "neurofibrils,” which are elements of the cytoskeleton stained with a silver solution [10,11].
Theoretical Framework
What Is the Blood–Brain Barrier?
The blood–brain barrier (BBB) is a complex, semi-permeable, and highly specialized interface composed of multiple cell types. Its primary function is to preserve the stability of the brain’s internal environment by restricting the passage of potentially harmful substances from the bloodstream into the central nervous system (CNS) [1,2]. This selective barrier poses a major challenge for the treatment of neurological diseases, as it also hinders the delivery of many therapeutic agents into brain tissue, including those intended for neurodegenerative conditions and brain malignancies [3].
Because neuronal cells are located extremely close to brain capillaries—at distances typically under 25 micrometers—the BBB represents a strategic and efficient route for targeted drug delivery, as opposed to longer and less effective systemic routes [10]. This proximity has encouraged scientific exploration into novel methods that can transiently modulate BBB permeability or bypass its restrictions altogether. Researchers are also focusing on how various delivery systems interact with the BBB and what structural features govern its selective permeability [3,4,5].
Composition of the Blood–Brain Barrier
The structural integrity of the BBB depends on several key proteins that form intercellular junctions. Among them, claudins, occludins, and junctional adhesion molecules play a central role in sealing the endothelial lining. These are accompanied by cytoplasmic scaffold proteins such as ZO-1, ZO-2, ZO-3, and cingulin, all of which interact with the actin cytoskeleton to support barrier function [12]. Claudins—particularly claudin-1 and claudin-5—along with occludin, are essential components of tight junctions. Occludin, a phosphoprotein, works in concert with other adhesion molecules, though the full extent of their function within the BBB remains under investigation [16]. Overall, the BBB is sustained by the coordinated efforts of endothelial cells, astrocytes, pericytes, and the complex junctional systems they form [13,15].
- - Endothelial Cells
Endothelial cells form the primary anatomical structure of the BBB, lining the cerebral vasculature and engaging in intricate communication with other CNS cell types. Unlike peripheral endothelial cells, those in the brain are characterized by tight and adherens junctions, which form sealed membrane compartments that reduce paracellular diffusion [20]. These cells lack fenestrations—small transcellular openings found in peripheral vasculature—which further enhances their barrier properties [21,22]. They also contain a higher number of mitochondria, reflecting the high metabolic demand required to sustain active transport processes. Functionally, BBB endothelial cells exhibit a net negative surface charge, reducing permeability to negatively charged molecules. They also express minimal leukocyte adhesion molecules, which limits immune cell infiltration. Moreover, specialized transporters within these cells tightly regulate the influx and efflux of substrates [17]. Their high transendothelial electrical resistance restricts the formation of transcellular vesicles, contributing to overall BBB impermeability [18,19].
- - Astrocytes
Astrocytes, or astroglial cells, are highly diverse and morphologically complex glial cells that play a fundamental role in CNS homeostasis. They are generally classified into protoplasmic types—found predominantly in the gray matter—and fibrous types, which are more common in the white matter. Astrocytes extend end feet that anchor to the capillary basement membrane via molecular complexes involving aquaporin-4, dystroglycan–dystrophin, and the proteoglycan agrin [23]. These cells contribute to several critical processes including synaptic signaling, clearance of extracellular waste, regulation of blood flow, ion homeostasis, and neuroimmune modulation [23,24]. However, the extent of their direct involvement in BBB formation remains debated. Some studies suggest astrocytes may induce barrier-like behaviors in endothelial or epithelial cells, whereas others propose the BBB emerges prior to astrocyte development [25]. Regardless, astrocytes, in tandem with pericytes, wrap around the cerebral vasculature and serve as key intermediaries between neurons and endothelial cells. Interestingly, in species lacking vascular systems, astrocyte-like cells constitute the main barrier separating hemolymph from the CNS [26,27].
- - Pericytes
Pericytes are mural cells that reside along the walls of capillary vessels and are embedded within the vascular basement membrane, situated just outside the endothelium. In the CNS, pericytes have near-complete coverage of the endothelial surface and are indispensable to BBB functionality. These cells engage in constant bidirectional communication with endothelial cells, exemplified by the PDGF-B/PDGFRβ signaling axis that facilitates pericyte recruitment and vessel stabilization [20,27]. Pericytes also modulate endothelial barrier properties by influencing tight junction protein expression and astrocyte polarization. Loss or dysfunction of pericytes compromises these interactions, leading to increased BBB permeability. Beyond structural support, pericytes regulate cerebral blood flow, contribute to angiogenesis, and participate in neuroinflammatory responses [17,23,28].
- - Tight Junctions
Tight junctions are essential for establishing the selective permeability of the BBB. These junctional complexes seal the paracellular space between endothelial cells, thereby limiting the diffusion of hydrophilic and high-molecular-weight substances. They act as both “gates”—regulating substance passage—and “fences”—preserving membrane polarity. Tight junctions are composed of transmembrane proteins such as claudins and occludins, which connect adjacent cells and are anchored to the cytoskeleton via adaptor proteins like ZO-1. Additional elements such as tricellulin, lipolysis-stimulated lipoprotein receptors, and adhesion molecules contribute to the maturation and functionality of tight junctions [31,33]. Kinases and other regulatory proteins further influence tight junction permeability, and any downregulation in their expression can lead to BBB disruption.
- - Adherent Junctions
Adherens junctions complement tight junctions by maintaining endothelial cohesion and supporting structural integrity. These microdomains are composed of vascular endothelial (VE)-cadherin and cytoplasmic catenins, which link transmembrane adhesion complexes to the actin cytoskeleton. VE-cadherin, in particular, mediates homophilic interactions between endothelial cells and stabilizes cell-cell adhesion. Catenins, p120 proteins, and placoproteins act as scaffolds that bridge adherens and tight junctions via ZO-1. Other molecules such as PECAM-1, CD99, and nectins also participate in adherens junction organization. Disruption of these complexes compromises barrier integrity and may lead to vascular leakage.
Function of the Blood-Brain Barrier
1.It protects the brain from circulating compounds and molecules in the bloodstream because of the tightly woven endothelium of the brain capillaries, which allows only oxygen, glucose, amino acids, and other essential nutrients to cross the BBB.
2.Selective transport from the capillary network to the brain parenchyma occurs by facilitated transport, as occurs with glucose, or by active diffusion that depends on ATP.
3. The BBB metabolizes or modifies the elements that pass from the blood to the nervous tissue and vice versa [17].
Transport Mechanisms Through the Blood–Brain Barrier
Brain capillary endothelial cells differ significantly from their peripheral counterparts due to the abundance of tight junctions, scarcity of vesicular transport, and elevated mitochondrial density. These tight junctions not only restrict paracellular permeability but also diversify the membrane compositions on opposite sides of the endothelial layer [12]. To overcome the restrictive nature of the BBB, various specialized transport systems have evolved to facilitate the controlled entry of molecules into the central nervous system [13].
- - Passive Diffusion
Small molecules may cross the BBB through either paracellular gaps or by transcellular routes. However, only a limited range of hydrophilic molecules can diffuse passively between tight junctions. In contrast, lipophilic compounds—such as ethanol and steroid hormones—can dissolve within the lipid bilayer of endothelial membranes and diffuse directly into the brain. For most essential nutrients, such as glucose and amino acids, passive diffusion is insufficient, necessitating more selective transport mechanisms [14].
- - Carrier-Mediated Transport
Integral membrane transporters enable specific nutrients and metabolites to cross the BBB efficiently. These proteins recognize and bind to their respective solutes—such as glucose or amino acids—and undergo structural shifts that ferry the molecules from regions of higher to lower concentration. When movement against a concentration gradient is required, energy in the form of ATP is employed, especially for charged or polar molecules [25,32,43].
- - Active Efflux Mechanisms
Efflux transporters actively remove both endogenous toxins and exogenous compounds, including therapeutic agents, from the brain into the bloodstream. A critical group in this function is the ATP-binding cassette (ABC) transporter family, which significantly influences drug pharmacokinetics within the central nervous system. While these pumps protect the brain from harmful agents, they also present a barrier to effective pharmacological treatment of neurological conditions [25].
- - Receptor-Mediated Transcytosis (RMT)
Receptor-mediated transcytosis enables selective transport of macromolecules via interaction with specific endothelial cell surface receptors. Molecules such as insulin, transferrin, and apolipoproteins bind to their respective receptors located in clathrin-coated pits of the luminal membrane. These invaginations form vesicles that internalize the ligand-receptor complex. After acidification within endosomes, the ligand may be released and transported across to the opposite side of the endothelial cell layer [16].
- - Adsorptive-Mediated Transcytosis (AMT)
Adsorptive-mediated transcytosis, often referred to as non-specific pinocytosis, involves the electrostatic interaction between cationic molecules and the negatively charged luminal surface of endothelial cells. Following this interaction, the bound molecule is internalized and transported through the endothelial cytoplasm, supported by the abundant mitochondria found within these cells, and eventually exocytosed on the abluminal side [17].
- - Cell-Mediated Transcytosis (CMT)
Cell-mediated transcytosis operates through the so-called “Trojan horse” mechanism, in which immune cells—such as monocytes or macrophages—carry various molecules across the BBB. This method enables transport of a broader range of substances, including those that cannot otherwise traverse the barrier. For instance, HIV-infected monocytes exploit this mechanism to infiltrate the brain. Emerging evidence also supports the use of CMT in therapeutic delivery, particularly in targeting drugs to the CNS [18].
Circumventricular Organs and the BBB
It is worth mentioning that there are also areas in the brain whose capillaries have fenestrated endothelium, that is, they do not have a BBB with consequent free exchange of molecules between the blood and neurons. These sites are mainly located around the ventricular cavities, and are therefore called circumventricular organs. These structures include the lamina terminalis vascularis organ (LTO), subfornical organ (SFO), subcommissural organ (SCO), median eminence, pineal gland, neurohypophysis, and the area postrema (AP). It is worth mentioning that choroid plexuses have been excluded from the list of circumventricular organs, although the reason for this is not clearly explained. [40]
Mechanisms of Blood-Brain Barrier Breakdown
Several factors contribute to BBB dysfunction. Neuroinflammation, mediated by cytokines such as IL1β and TNFα, disrupts tight junctions [10]. Vascular injury due to ischemia increases permeability through VEGF and nitric oxide pathways [11]. Oxidative stress damages endothelial cells and junctional proteins [12]. Activated microglia facilitate leukocyte infiltration through the BBB, exacerbating CNS injury [13].
Blood–Brain Barrier Disruption and Its Role in Neurological Disorders
A variety of pathological conditions, including infections and traumatic injuries, can compromise the structural and functional integrity of the blood–brain barrier (BBB) by damaging both the tight junctions and the neurovascular unit (NVU). When this finely regulated barrier is disrupted, its capacity to control the selective exchange of ions and molecules between the blood and brain tissue is significantly impaired [5]. As a result, harmful agents such as pathogens, immune cells, or toxins may infiltrate the central nervous system, triggering neuroinflammatory responses. Inflammatory mediators, including cytokines released in response to immune activation, can promote neuronal dysfunction and degeneration, ultimately contributing to the onset or progression of neurological diseases [6].
The BBB is composed of highly specialized endothelial cells that interact closely with astrocytic endfeet. These astrocyte processes surround cerebral microvessels in rosette-like formations, playing essential roles in maintaining the ionic and osmotic balance of the neural environment. They regulate water and potassium homeostasis through specific channels, including aquaporin-4 and Kir4.1. Astrocytes also secrete signaling molecules such as sonic hedgehog (Shh) and basic fibroblast growth factor (bFGF), which modulate endothelial phenotype and enhance the expression of tight junction proteins—occludin, claudins, and zonula occludens (ZO)—reinforcing barrier selectivity and cohesion [20,26]. Additionally, astrocyte-derived α-dystrobrevin contributes to barrier stability by promoting cytoskeletal organization and cell–cell adhesion.
Through the combined action of these regulatory pathways, astrocytes not only sustain BBB architecture but also help limit the passage of hydrophilic or potentially harmful substances from the systemic circulation into the brain. Molecules such as bFGF, transforming growth factor-β (TGF-β), and various neurotrophic factors further reinforce BBB impermeability by enhancing the expression of selective transporters and tight junction components. This contributes to an active exchange system that restricts passive diffusion of polar molecules while enabling the import of essential nutrients and the export of metabolic waste.
Compared with vascular endothelial cells in other organs, those within the brain express significantly higher levels of occludin, claudin-5, and ZO-1. This specialization is essential for preserving central nervous system homeostasis [28].
Alzheimer’s Disease
Alzheimer’s disease (AD), the most prevalent neurodegenerative condition globally, is marked by hallmark pathological features such as extracellular beta-amyloid (Aβ) deposits and tau protein aggregates forming neurofibrillary tangles. Alongside these lesions, increasing evidence highlights concomitant vascular abnormalities. These may include damage to cerebral microvessels, reduced cerebral perfusion, and perivascular accumulation of Aβ peptides. Notably, vascular impairment is now recognized as an early contributor to AD pathology and may exacerbate both cognitive decline and the progression of neuronal damage [38].
The Blood-Brain Barrier and Alzheimer's Disease
It is common knowledge that it was long considered a disease exclusive to the elderly and has begun to be detected in very young patients. It has been proven that addictions, such as alcoholism, smoking, and drug addiction, can cause its early onset, since certain substances enter the bloodstream and, due to their high toxicity, alter the behavior of the BBB, reach neurons, and alter them.
To corroborate this, contrast-enhanced brain MRI scans were performed, and the images showed that the blood-brain barrier that protects the brain developed leaks with age. These leaks begin in the hippocampus, an important learning and memory center that is damaged in Alzheimer's disease. Postmortem examination of the brains of patients with Alzheimer's disease has also revealed damage to the blood-brain barrier. [39]
Comparison Between the Causes of Alzheimer's Disease and Blood-Brain Barrier Impairment
- Function of microglia
-Alzheimer's disease (AD)
Insights from genome-wide association studies (GWAS) have underscored the critical involvement of microglial immune mechanisms in the pathophysiology of Alzheimer’s disease (AD). Several genes identified as risk alleles—most notably TREM2 and APOE—are predominantly expressed in microglial cells, suggesting these cells play a central role in disease susceptibility.
Contrary to earlier hypotheses that emphasized a classic pro-inflammatory microglial response, postmortem analyses of AD patients have revealed the presence of morphologically distinct microglial phenotypes surrounding amyloid-β (Aβ) deposits and tau aggregates. These cells exhibit features that align more closely with dystrophic or senescent microglia, rather than with the activated states typically observed in acute inflammatory contexts such as sepsis. This has led to the proposal that microglial dysfunction in AD contributes more to synaptic degradation and neuronal loss than to inflammatory damage per se [48,49].
In physiological contexts, microglia play a pivotal role during neurodevelopment, eliminating neurons that fail to integrate into functional networks and actively remodeling synaptic connections. This pruning process is mediated through the classical complement cascade, wherein synaptic targets are tagged with C1q and C3 proteins and subsequently recognized by complement receptor 3 (C3R) on microglia. The removal occurs via trogocytosis, a process that selectively targets presynaptic terminals and axonal segments. These mechanisms are particularly relevant to AD, where synaptic loss and cognitive decline accompany the accumulation of pathological protein aggregates, implicating aberrant microglial activity in disease progression [45].
-Blood–Brain Barrier
Under normal conditions, microglial activity is tightly regulated by the intact blood–brain barrier (BBB), which prevents direct exposure to circulating blood components. However, in the setting of acute injury or neurodegeneration, BBB disruption allows plasma-derived molecules to infiltrate the brain parenchyma, activating microglial responses. To investigate the specific impact of circulating plasma factors, researchers introduced plasma from wild-type mice into the corpus callosum of recipient animals. The result was a significant shift in microglial gene expression, particularly in pathways associated with cytoskeletal reorganization, oxidative phosphorylation, stress response, and transcriptional control.
Interestingly, when plasma from fibrinogen alpha chain-deficient mice (Fga-/-) was used, a marked downregulation of genes related to oxidative stress (e.g., Hmox1, Cox7a2, Slc25a5) and disease-associated microglial profiles (e.g., Ccl12, Rps8, Rpl35, Atp5e, Psmd2, Tubb5) was observed. These findings point to fibrinogen—a key coagulation protein—as a potent activator of microglia, particularly in the context of cerebrovascular events such as stroke or minor hemorrhage [43,44].
Red blood cells (RBCs), when exposed to oxidative insults, can also provoke microglial activation and impair cerebral microcirculation. In experiments using Tie2-GFP transgenic mice—which express fluorescent markers in endothelial cells—oxidatively stressed RBCs (labeled with PKH26 and treated with t-butyl hydroperoxide) were injected to simulate a redox challenge. Compared with controls injected with phosphate-buffered saline (PBS), these animals showed increased vascular stasis and reduced cerebral blood flow within 1 to 24 hours post-injection, as visualized through two-photon imaging [50].
Additionally, perivascular microglia—distinct from resident parenchymal microglia—derive from bone marrow precursors and localize to the vasculature. These macrophage-like cells are strategically positioned to mediate immune surveillance and may play a central role in BBB dysfunction during autoimmune and inflammatory diseases, particularly those involving leukocyte transmigration across the endothelium [25].
- Inflammation
-Alzheimer
Data from postmortem analyses of Alzheimer’s disease (AD) brains—obtained through immunohistochemical, biochemical, and molecular techniques—have made it possible to classify the wide array of inflammatory molecules found within the central nervous system (CNS) of affected individuals. Central to the inflammatory cascade in AD are β-amyloid (Aβ) deposits, tau-based neurofibrillary tangles, and progressive neuronal degeneration—hallmark lesions that have been recognized for nearly a century [51]. Similar to peripheral tissues, where chronic injury and persistent exposure to aberrant materials trigger immune activation, the accumulation of Aβ and tau in the brain is believed to initiate a sustained inflammatory response. Once this is set in motion, a multitude of immune pathways become involved, with overlapping feedback loops and intricate cross-talk between mediators [32,42].
This complexity makes it likely that the activation of one inflammatory axis can influence many others. Therefore, any attempt to identify a primary driver of inflammation in AD is more a matter of convenience than definitive hierarchy. Whether focusing on complement activation, cytokine release, chemokine signaling, acute-phase reactants, or other pathways, current evidence suggests that no single mechanism outweighs the others in its contribution to disease progression [20,33,50,53].
Epidemiological studies have reported a decreased risk of developing AD in individuals chronically treated with anti-inflammatory medications for conditions such as arthritis. This supports the hypothesis that inflammation plays a significant role in AD pathology. Inflammation, as an innate immune reaction to injury, infection, or systemic disorders—including obesity—can be acute or chronic [24]. While acute inflammation presents with clear clinical signs such as fever, pain, and swelling, chronic inflammation often persists silently and may underlie neurodegenerative processes. In the AD brain, inflammatory cells and mediators accumulate in close proximity to Aβ plaques. Once initiated, the inflammatory cascade leads to the release of cytotoxic factors that compromise cellular membranes, promoting neuronal lysis and the spread of damage across broader regions of the brain [2,15].
-Blood-brain barrier
Neuroinflammatory responses in AD are frequently accompanied by alterations in the cerebral microvasculature. Notably, systemic infections that do not directly affect the CNS can still trigger brain inflammation, suggesting that peripheral immune activity contributes to the neuroinflammatory milieu. Although the BBB serves as a gatekeeper between the bloodstream and the CNS, endothelial cells are not passive participants. Instead, they actively transmit inflammatory signals to adjacent perivascular macrophages and microglia, which may in turn affect neurons and glial cells within the brain parenchyma.
Key proinflammatory cytokines—such as interleukin-6 (IL-6), tumor necrosis factor-alpha (TNF-α), and interleukin-1 beta (IL-1β)—can traverse the BBB through saturable, receptor-mediated transport systems [29]. The efficiency of transport depends on both the molecular structure of the cytokine and its plasma concentration. Some cytokines are internalized by endothelial cells, while others penetrate deeper into the CNS tissue. Interestingly, studies have demonstrated that plasma from elderly individuals can enhance the expression of vascular cell adhesion molecule-1 (VCAM-1) in cerebral endothelial cells. This upregulation promotes microglial activation and facilitates the adhesion and migration of leukocytes, reflecting endothelial activation and immune infiltration [40,41,43].
From an immunological perspective, it is now well established that CNS inflammation increases the permeability of the BBB, especially in the presence of antigenic stimulation. This heightened permeability allows the infiltration of T lymphocytes and neutrophils into the brain, a hallmark of neuroinflammatory activity. The molecular pathways leading to this barrier disruption involve complex interactions between inflammatory mediators and perivascular cells—particularly microglia—which play a pivotal role in modulating endothelial integrity and facilitating immune cell transmigration [45].
- Vascular damage
-Alzheimer
Vascular dysfunction in Alzheimer’s disease (AD) creates conditions where circulating blood components and neural proteins can interact in harmful ways. One of the clearest examples of this pathological interface is cerebral amyloid angiopathy (CAA) [3,30], a condition marked by the deposition of amyloid-β (Aβ) within and around the cerebral vasculature. CAA is observed in approximately 80–95% of individuals with AD and is associated with endothelial injury, vascular instability, and inflammatory processes. As a consequence, patients with CAA present a higher risk of experiencing cerebrovascular events, particularly stroke, compared to those without vascular amyloid deposition [30,44]. Additionally, inherited variants of CAA have been linked to altered neural network connectivity, indicating that vascular compromise may directly contribute to cognitive decline [54]. The compromised vascular barrier in CAA enhances the interaction between Aβ and plasma proteins like fibrinogen, a key factor in coagulation. When Aβ binds to fibrinogen, it alters fibrin clot architecture and impairs fibrinolysis, thus exacerbating vascular dysfunction [22,24]. Specific mutations in Aβ, particularly those linked to early-onset AD, show stronger affinities for fibrinogen, further increasing amyloid accumulation within the vasculature and intensifying endothelial damage [23]. Compounding this, neuroinflammation has been identified as a major contributor to vascular and neural deterioration in AD. Autopsy reports have consistently shown elevated inflammatory markers [26], and neuroimaging has revealed persistent inflammation across the disease course [24,25,26].
Moreover, growing evidence supports a connection between cardiovascular disease and the risk of developing Alzheimer’s disease. As early as 1969, studies reported increased levels of homocysteine (Hcy)—a byproduct of protein metabolism—in the urine of patients with cardiovascular pathology. Elevated Hcy in circulation can contribute to vascular damage by promoting the oxidation of cholesterol and compromising endothelial function [52]. These vascular insults may serve as a common mechanistic link between systemic cardiovascular risk factors and neurodegeneration observed in AD.
-Blood-Brain Barrier
Alterations in cerebral blood flow in Alzheimer’s disease (AD) exacerbate vascular injury, a process further intensified by the breakdown of the blood–brain barrier (BBB). Emerging research attributes this deterioration, in part, to the degeneration of pericytes, contractile cells that enwrap the endothelium and contribute to BBB stability [27,32,37,46,52]. Neuroimaging has revealed that the hippocampus, a region essential for memory, is particularly susceptible to pericyte-related barrier dysfunction, with this vulnerability increasing as individuals age [28]. Transcriptomic analyses using VINE-seq (vessel isolation and nuclear extraction for sequencing) in human brain tissue have identified multiple vascular alterations in patients with AD compared to cognitively healthy controls [18,53]. The reduced number of recovered vascular cell nuclei in AD samples, validated by immunostaining, suggests cell loss within the cerebral vasculature. Additionally, significant changes in gene expression were observed in mural cells and fibroblasts, both of which play essential roles in maintaining vascular tone and barrier architecture. These molecular alterations imply impaired vasoconstriction and disrupted cerebral perfusion, potentially explaining some of the cerebrovascular abnormalities detected in AD [54].
Hypoxic and ischemic events are closely associated with the disruption of endothelial junctions that maintain BBB integrity. This breakdown is mediated by key molecular players, including inflammatory cytokines, vascular endothelial growth factor (VEGF), and nitric oxide (NO). In particular, elevated concentrations of interleukin-1β (IL-1β) and tumor necrosis factor-alpha (TNF-α) have been reported in such conditions. These mediators enhance the expression of adhesion molecules on endothelial cells, as well as on circulating immune cells like monocytes and neutrophils, facilitating their transmigration across the BBB. As a result, leukocyte infiltration into the brain parenchyma increases. While these inflammatory cascades contribute to barrier damage, astrocytes may act as modulators of injury, playing a protective role by regulating vascular responses during such insults [55].
- Oxidative stress
-Alzheimer
Oxidative stress is a widely recognized contributor to neurodegenerative conditions, including Alzheimer’s disease (AD). Both oxidative imbalance and the aggregation of amyloid-β (Aβ) oligomers are events that occur prior to the manifestation of clinical symptoms in AD. Research using transgenic mouse models carrying AD-related genetic mutations has consistently shown elevated levels of oxidative damage markers [4,14,37]. The brain is especially susceptible to oxidative injury due to several intrinsic factors: its high demand for oxygen, abundance of polyunsaturated fatty acids, limited antioxidant defenses, and the presence of transition metals capable of redox cycling. The development of animal models expressing key mutations associated with AD has facilitated the investigation of oxidative stress in the early phases of the disease. Notably, biochemical signs of oxidative damage—particularly those linked to lipid peroxidation—can be detected before the formation of Aβ plaques and cognitive decline, making them promising indicators for early diagnosis [44]. Common biomarkers of oxidative damage include lipid peroxidation products such as F2-isoprostanes (F2-IsoPs), F4-neuroprostanes (F4-NeuroPs), malondialdehyde (MDA), and 4-hydroxy-2-nonenal (HNE) [25,33,40,45]. In addition, protein oxidation is evidenced by increased carbonylation, while oxidative injury to nucleic acids is reflected in elevated levels of 8-oxo-2’-deoxyguanosine (8-OHdG) and 8-hydroxyguanosine (8-OHG), which serve as key markers of DNA and RNA damage, respectively [50].
-Blood–Brain Barrier
The degradation of epithelial and endothelial barriers is a key event in the development of inflammation following oxidative stress. Endothelial cells serve both as initiators and as targets of reactive oxygen species (ROS), which contribute to vascular injury by modifying lipids and proteins within affected cells. This oxidative damage triggers the activation and release of inflammatory mediators, including cytokines and proteolytic enzymes, which in turn amplify tissue injury. In the context of acute ischemic stroke, the most extensively studied pro-inflammatory cytokines include tumor necrosis factor-alpha (TNF-α), various interleukins (IL-1β, IL-6, IL-10, IL-17, IL-20), and transforming growth factor-beta (TGF-β), all of which play pivotal roles in orchestrating vascular and immune responses [52,55]. Among these, interleukin-17A (IL-17A) has been shown to stimulate ROS production via NADPH oxidase or xanthine oxidase pathways. The resulting oxidative stress enhances the activity of endothelial contractile proteins and leads to reduced expression of occludin, a tight junction protein essential for blood–brain barrier integrity. Notably, inhibiting either ROS generation or myosin light chain phosphorylation has been found to prevent IL-17A-induced disruption of the BBB [27,30,47].
As detailed in Supplementary Table 1, these mechanisms share a close functional and structural relationship between BBB disruption and the progression of Alzheimer’s disease.
In addition to inflammation, oxidative stress, vascular injury, and impaired amyloid-β clearance, other mechanisms reinforce the interconnection between blood-brain barrier (BBB) dysfunction and Alzheimer's disease (AD) pathogenesis.
- -
- Pericyte degeneration: Pericytes are perivascular cells crucial for the stability and function of the blood-brain barrier (BBB). Their degeneration, observed in the early stages of AD, contributes to the loss of the blood-brain barrier, cerebral hypoperfusion, and impaired neuronal homeostasis [27,28]. Murine models have demonstrated that pericyte loss is directly associated with white matter damage and cognitive deficits [50].
- -
- Nutrient transport dysfunction: The glucose transporter GLUT1, which is essential for brain energy supply, is decreased in the blood-brain barrier (BBB) of patients with AD [29]. This metabolic dysfunction compromises neuronal viability, facilitates the accumulation of pathological proteins, and accelerating neurodegeneration [15,16].
- -
- Loss of polarity in astrocytes: Astrocytes normally regulate metabolic exchange and blood-brain barrier (BBB) integrity through their perivascular terminals. In AD, the loss of polarity in astrocytes alters their supportive functions, increasing vascular permeability and exacerbating neuroinflammation [5,8].
- -
- -
- Endothelial epigenetic modifications: Epigenetic alterations in brain endothelial cells, driven by chronic inflammation and aging, reduce the expression of essential tight junction proteins and transporters and promote the disruption of the blood-brain barrier (BBB) [26]. These modifications could represent an early link between environmental risk factors and susceptibility to AD [56].
- -
- Dysregulation of the Wnt/β-catenin pathway: The Wnt/β-catenin signaling pathway is crucial for the maintenance of the blood-brain barrier (BBB). Its inhibition, observed in AD, leads to decreased expression of tight junction proteins and structural weakening of the barrier [29].
- -
- Hormonal stress: mild stress, chronically experienced, aggravates, and accelerates the main features of the disease in people with a genetic predisposition to develop Alzheimer's disease. Many studies have shown that stress can cause cognitive impairments. In addition, patients with depression experience episodes of memory loss, and stress is a factor associated with depression [17,46,49]. Stress and hormones released during stress exposure can also alter the normal functioning of the BBB, since most cells involved in BBB formation (endothelial cells, astrocytes, and microglia) have receptors for glucocorticoids, corticotropin-releasing hormone, and adrenaline. In adult mammals, acute stress modifies BBB permeability to circulating molecules in the blood, and several studies have reported an increase in BBB permeability after acute stress [25].
These additional mechanisms reinforce the hypothesis that blood-brain barrier (BBB) dysfunction is not simply a secondary event but a fundamental and early contributor to the Alzheimer's disease cascade. Therefore, therapeutic strategies aimed at preserving BBB integrity have the potential to delay or prevent neurodegeneration [47,48].
What Starts Outside the Organism (Parabiosis)
The beta-amyloid protein performs numerous basic functions in the body. However, when they acquire the wrong structure, these proteins stick together, forming fibers that, in turn, aggregate into oligomers and beta-amyloid plaques, which are highly toxic to neurons. Numerous studies have suggested that these plaques are responsible for brain cell death that triggers Alzheimer's disease. However, this protein is not unique to the brain but is produced throughout the body [4,12]. Therefore, is it possible that beta-amyloid protein of "extracerebral" origin also contributes to the onset and progression of Alzheimer's disease?
To answer this question, the authors resorted to a technique called "parabiosis,” in which two living beings are surgically joined to form, as if they were Siamese twins, a "single organism" that shares the physiological systems of its two predecessors. They used two mice and "stitched" them together to create a "single mouse" with a shared circulatory system. The first mouse was completely normal and, therefore, unable to develop Alzheimer's disease. However, the second mouse was genetically modified to carry a mutation responsible for the production of high levels of beta-amyloid protein. Thus, after several months, a large number of beta-amyloid plaques were observed in the brain. At this point, he was stitched together with his healthy counterpart for one year so that the brains of both animals ended up sharing the disease [9].
Results
Recent findings support the notion that Alzheimer’s disease (AD) arises not solely from central nervous system dysfunction but also from systemic processes contributing to the accumulation of amyloid-β (Aβ). This perspective opens therapeutic opportunities that extend beyond the brain, such as peripheral interventions involving liver and kidney clearance of circulating Aβ through targeted molecules capable of binding and facilitating its elimination [48,49].
Our histological analysis of the blood–brain barrier (BBB) reinforces the hypothesis that increased permeability of this structure may represent a crucial early event in AD pathogenesis. The BBB is not simply a passive protective barrier but a dynamic interface highly sensitive to many of the same pathogenic factors implicated in AD—oxidative stress, inflammation, vascular dysfunction, and aging. However, a paradox emerges: while the BBB permits the entry of harmful agents under pathological conditions, it often prevents the passage of therapeutic compounds intended to restore brain homeostasis. This raises an urgent and unresolved question: what distinguishes the physicochemical properties of noxious substances that cross the BBB from those of drugs designed to benefit the brain? Is the selectivity of a damaged BBB altered in such a way that it favors pathology over repair? These are challenges that demand deeper investigation.
Advances in drug delivery strategies, including functionalized nanoparticles, have begun to overcome these obstacles by enabling targeted and controlled delivery of therapeutic agents across the BBB to sites of neurodegeneration [8]. Moreover, preventive strategies focused on restoring or resealing BBB integrity are gaining attention, as chronic exposure to blood-borne toxins is increasingly seen as a driver of neurodegeneration [56]. Stress, as a modifiable risk factor, may also influence both BBB permeability and AD vulnerability. While glucocorticoid-mediated stress responses vary among individuals, preclinical models suggest that those with higher stress resilience may exhibit resistance to AD pathology [52,55,56]. These findings suggest that the future of AD prevention and treatment lies not only in targeting neural circuits, but also in addressing systemic, vascular, immunological, and behavioral factors that converge at the blood–brain interface.
Conclusions
The breakdown of the blood–brain barrier (BBB) is increasingly recognized not merely as a consequence but as a central, active contributor to the pathogenesis of Alzheimer’s disease (AD). Far from being a passive filter, the BBB plays a dynamic regulatory role in maintaining central nervous system (CNS) homeostasis through selective transport, immune surveillance, and neurovascular coupling. Its structural integrity—maintained by endothelial cells, pericytes, astrocytic endfeet, and extracellular matrix components—is progressively compromised in AD due to a multifactorial convergence of amyloid pathology, tau-related cytotoxicity, chronic neuroinflammation, oxidative stress, and vascular dysfunction.
The evidence reviewed suggests that BBB disruption precedes cognitive symptoms and facilitates a vicious cycle: impaired clearance of amyloid-β, increased infiltration of peripheral inflammatory mediators, dysregulated cerebral perfusion, and enhanced oxidative damage. Cerebral amyloid angiopathy, pericyte degeneration, tight junction downregulation, and the aberrant activation of microglia all converge at the interface between vascular and neurodegenerative processes. This intersection reveals that AD should be reconsidered, at least in part, as a neurovascular disorder.
Despite these advances, current therapeutic approaches largely overlook the vascular and barrier-related aspects of AD. Targeting BBB preservation—whether by modulating endothelial signaling, stabilizing pericyte function, inhibiting pro-inflammatory cascades, or restoring transporter systems—offers an underexplored but potentially transformative avenue for early intervention and disease modification. Future research should aim not only to characterize BBB dysfunction as a biomarker of early AD but also to exploit it as a therapeutic entry point. Understanding and restoring BBB function may ultimately prove essential for altering the course of neurodegeneration in Alzheimer’s disease.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org.
Author Contributions
All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
The study was conducted in accordance with the Declaration of Helsinki, and approved by the Institutional Review Board.
Informed Consent Statement
Not applicable.
Acknowledgments
The authors have reviewed and edited the output and take full responsibility for the content of this publication.
Conflicts of Interest
The authors declare that there are no conflicts of interest regarding the publication of this paper.
References
- Lane, C.A.; Hardy, J.; Schott, J.M. Alzheimer's disease. Eur J Neurol 2018, 25, 59–70. [Google Scholar] [CrossRef] [PubMed]
- Selkoe, D.J.; Hardy, J. The amyloid hypothesis of Alzheimer's disease at 25 years. EMBO Mol Med 2016, 8, 595–608. [Google Scholar] [CrossRef] [PubMed]
- Zlokovic, B.V. The blood–brain barrier in health and chronic neurodegenerative disorders. Neuron 2008, 57, 178–201. [Google Scholar] [CrossRef] [PubMed]
- Abbott, N.J.; Rönnbäck, L.; Hansson, E. Astrocyte–endothelial interactions at the blood–brain barrier. Nat Rev Neurosci 2006, 7, 41–53. [Google Scholar] [CrossRef] [PubMed]
- Obermeier, B.; Daneman, R.; Ransohoff, R.M. Development, maintenance and disruption of the blood–brain barrier. Nat Med 2013, 19, 1584–1596. [Google Scholar] [CrossRef] [PubMed]
- Nation DA, Sweeney MD, Montagne A, et al. Blood–brain barrier breakdown is an early biomarker of cognitive dysfunction. Nat Med 2019, 25, 270–276. [Google Scholar] [CrossRef] [PubMed]
- Montagne A, Nikolakopoulou AM, Zhao Z, et al. Blood–brain barrier breakdown in the aging human hippocampus. Neuron 2020, 85, 296–302. [Google Scholar]
- Zlokovic, B.V. Neurovascular pathways to neurodegeneration in Alzheimer's disease and other disorders. Nat Rev Neurosci 2011, 12, 723–738. [Google Scholar] [CrossRef] [PubMed]
- Deane, R.; Sagare, A.; Zlokovic, B.V. The role of the cell surface LRP and soluble LRP in blood–brain barrier Aβ clearance in Alzheimer's disease. Curr Pharm Des 2008, 14, 1601–1605. [Google Scholar] [CrossRef] [PubMed]
- Heneka, M.T.; Golenbock, D.T.; Latz, E. Innate immunity in Alzheimer's disease. Nat Immunol 2015, 16, 229–236. [Google Scholar] [CrossRef] [PubMed]
- Iadecola, C. The pathobiology of vascular dementia. Neuron 2013, 80, 844–866. [Google Scholar] [CrossRef] [PubMed]
- Butterfield, D.A.; Halliwell, B. Oxidative stress, dysfunctional glucose metabolism and Alzheimer disease. Nat Rev Neurosci 2019, 20, 148–160. [Google Scholar] [CrossRef] [PubMed]
- Ransohoff, R.M. How neuroinflammation contributes to neurodegeneration. Science 2016, 353, 777–783. [Google Scholar] [CrossRef] [PubMed]
- Montagne A, Nation DA, Sagare AP, et al. APOE4 leads to blood–brain barrier dysfunction predicting cognitive decline. Nature 2015, 581, 71–76. [Google Scholar]
- Sweeney, M.D.; Sagare, A.P.; Zlokovic, B.V. Blood–brain barrier breakdown in Alzheimer disease and other neurodegenerative disorders. Nat Rev Neurol 2018, 14, 133–150. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, Y.; Kanekiyo, T. Blood–brain barrier dysfunction and the pathogenesis of Alzheimer's disease. Int J Mol Sci 2017, 18, 1965. [Google Scholar] [CrossRef] [PubMed]
- Gorelick PB, Scuteri A, Black SE, et al. Vascular contributions to cognitive impairment and dementia. Stroke 2011, 42, 2672–2713. [Google Scholar] [CrossRef] [PubMed]
- Kalaria, R.N. The pathology and pathophysiology of vascular dementia. Neuropharmacology 2018, 134, 226–239. [Google Scholar] [CrossRef] [PubMed]
- Saraiva, C.; Praça, C.; Ferreira, R.; Santos, T.; Ferreira, L.; Bernardino, L. Nanoparticle-mediated brain drug delivery: overcoming blood–brain barrier to treat neurodegenerative diseases. J Control Release 2016, 235, 34–47. [Google Scholar] [CrossRef] [PubMed]
- Niu, X.; Chen, J.; Gao, C. Nanotechnology strategies for the diagnosis and therapy of Alzheimer's disease. Front Bioeng Biotechnol 2021, 9, 654994. [Google Scholar]
- Hansen, D.V.; Hanson, J.E.; Sheng, M. Microglia in Alzheimer's disease. J Cell Biol 2018, 217, 459–472. [Google Scholar] [CrossRef] [PubMed]
- Butterfield, D.A.; Boyd-Kimball, D. Oxidative stress, amyloid-β peptide, and altered key molecular pathways in the pathogenesis and progression of Alzheimer's disease. J Alzheimers Dis 2018, 62, 1345–1367. [Google Scholar] [CrossRef] [PubMed]
- Sultana, R.; Perluigi, M.; Butterfield, D.A. Oxidatively modified proteins in Alzheimer's disease and mild cognitive impairment. Acta Neuropathol 2009, 118, 131–150. [Google Scholar] [CrossRef] [PubMed]
- Kreuter, J. Drug delivery to the central nervous system by polymeric nanoparticles: what do we know? Adv Drug Deliv Rev 2014, 71, 2–14. [Google Scholar] [CrossRef] [PubMed]
- Erickson, M.A.; Banks, W.A. Blood–brain barrier dysfunction as a cause and consequence of Alzheimer's disease. J Cereb Blood Flow Metab 2013, 33, 1500–1513. [Google Scholar] [CrossRef] [PubMed]
- Zlokovic, B.V.; Sagare, A.P.; Sweeney, M.D. Blood–brain barrier breakdown in Alzheimer disease and other neurodegenerative disorders. Nat Rev Neurol 2018, 14, 133–150. [Google Scholar]
- Montagne, A.; Nikolakopoulou, A.M.; Zhao, Z.; et al. Pericyte degeneration causes white matter dysfunction in the mouse, C. N.S. Nat Med 2018, 24, 326–337. [Google Scholar] [CrossRef] [PubMed]
- Sweeney, M.D.; Ayyadurai, S.; Zlokovic, B.V. Pericytes of the neurovascular unit: key functions and signaling pathways. Nat Neurosci 2016, 19, 771–783. [Google Scholar] [CrossRef] [PubMed]
- Zhang X, Wang R, Chen S, et al. Blood–brain barrier dysfunction in Alzheimer's disease: pathogenic mechanisms and therapeutic targets. CNS Neurosci Ther 2024, 30, 5–19. [Google Scholar]
- Zhou Y, Wang Y, Jiang H, et al. The blood–brain barrier in Alzheimer's disease: A target for therapeutic intervention. Front Aging Neurosci 2023, 15, 1258640. [Google Scholar] [CrossRef] [PubMed]
- Fraser, P.E.; Nguyen, J.T.; McLachlan, D.R.; Abraham, C.R.; Kirschner, D.A. Alpha 1-antichymotrypsin binding to Alzheimer A beta peptides is sequence specific and induces fibril disaggregation in vitro. J Neurochem. 1993, 61, 298–305. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; He, Y.; Han, J.; Wei, W.; Chen, F. Blood–brain barrier dysfunction and Alzheimer's disease: associations, pathogenic mechanisms, and therapeutic potential. Front Aging Neurosci. 2023, 15, 1258640. [Google Scholar] [CrossRef] [PubMed]
- Preis, L.; Villringer, K.; Brosseron, F.; et al. Assessing blood–brain barrier dysfunction and its association with Alzheimer's pathology, cognitive impairment and neuroinflammation. Alzheimers Res Ther. 2024, 16, 172. [Google Scholar] [CrossRef] [PubMed]
- Lyu, Y.; Meng, Z.; Hu, Y.; et al. Mechanisms of mitophagy and oxidative stress in cerebral ischemia–reperfusion, vascular dementia, and Alzheimer's disease. Front Mol Neurosci. 2024, 17, 1394932. [Google Scholar] [CrossRef] [PubMed]
- Pizzino, G.; Irrera, N.; Cucinotta, M.; et al. Oxidative stress: harms and benefits for human health. Oxid Med Cell Longev. 2017, 2017, 8416763. [Google Scholar] [CrossRef] [PubMed]
- Song, K.; Li, Y.; Zhang, H.; et al. Oxidative stress-mediated blood–brain barrier disruption in neurological diseases. Oxid Med Cell Longev. 2020, 2020, 4356386. [Google Scholar] [CrossRef]
- Lehner, C.; Gehwolf, R.; Tempfer, H.; et al. Oxidative stress and blood–brain barrier dysfunction under particular consideration of matrix metalloproteinases. Antioxid Redox Signal. 2011, 15, 1305–1323. [Google Scholar] [CrossRef] [PubMed]
- Chung, T.D.; Linville, R.M.; Guo, Z.; et al. Effects of acute and chronic oxidative stress on the blood–brain barrier in 2D and 3D in vitro models. Fluids Barriers CNS. 2022, 19, 33. [Google Scholar] [CrossRef] [PubMed]
- Alcendor, D.J. Interactions between amyloid-β proteins and human brain pericytes: implications for the pathobiology of Alzheimer's disease. J Clin Med. 2020, 9, 1490. [Google Scholar] [CrossRef] [PubMed]
- Banks, W.A.; Rhea, E.M. The blood–brain barrier, oxidative stress, and insulin resistance. Antioxidants. 2021, 10, 1695. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, W.; Ijaz, B.; Shabbiri, K.; et al. Oxidative toxicity in diabetes and Alzheimer's disease: mechanisms behind ROS/RNS generation. J Biomed Sci. 2017, 24, 76. [Google Scholar] [CrossRef] [PubMed]
- Da, F.A.; Matias, D.; Garcia, C.; et al. The impact of microglial activation on blood–brain barrier in brain diseases. Front Cell Neurosci. 2014, 8, 362. [Google Scholar]
- Raas, Q.; Tawbeh, A.; Tahri-Joutey, M.; et al. Impaired peroxisomal beta-oxidation in microglia triggers oxidative stress and impacts neurons and oligodendrocytes. Front Mol Neurosci. 2025, 18, 1542938. [Google Scholar]
- Rojo, A.I.; McBean, G.; Cindric, M.; et al. Redox control of microglial function: molecular mechanisms and functional significance. Antioxid Redox Signal. 2014, 21, 1766–1801. [Google Scholar] [CrossRef] [PubMed]
- Tian, Z.; Ji, X.; Liu, J. Neuroinflammation in vascular cognitive impairment and dementia: current evidence, advances, and perspectives. Int J Mol Sci. 2022, 23, 6224. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Zhang, Z.; Li, H.; et al. Blockage of VEGF function by bevacizumab alleviates early-stage cerebrovascular dysfunction and improves cognitive function in a mouse model of Alzheimer's disease. Transl Neurodegener. 2024, 13, 1. [Google Scholar] [CrossRef] [PubMed]
- Velmurugan, G.V.; Vekaria, H.J.; Hartz, A.M.S.; et al. Oxidative stress alters mitochondrial homeostasis in isolated brain capillaries. Fluids Barriers CNS. 2024, 21, 81. [Google Scholar] [CrossRef] [PubMed]
- Fang, X.; Zhang, Y.; Zhang, Y.; et al. Endothelial extracellular vesicles: their possible function and clinical significance in diabetic vascular complications. J Transl Med. 2024, 22, 944. [Google Scholar] [CrossRef] [PubMed]
- Higashi, Y. Roles of oxidative stress and inflammation in vascular endothelial dysfunction-related disease. Antioxidants. 2022, 11, 1958. [Google Scholar] [CrossRef] [PubMed]
- Abbott, N.J.; Rönnbäck, L.; Hansson, E. Astrocyte-endothelial interactions at the blood–brain barrier. Nat Rev Neurosci. 2006, 7, 41–53. [Google Scholar] [CrossRef] [PubMed]
- Sofroniew, M.V. Astrocyte barriers to neurotoxic inflammation. Nat Rev Neurosci. 2015, 16, 249–263. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Zhao, Q.; Zhang, Y.; et al. A new andrographolide derivative ADA targeting SIRT3-FOXO3a signaling mitigates cognitive impairment by activating mitophagy and inhibiting neuroinflammation in Apoe4 mice. Phytomedicine. 2024, 124, 155298. [Google Scholar] [CrossRef] [PubMed]
- Zuo, M.L.; Wang, A.P.; Song, G.L.; Yang, Z.B. miR-652 protects rats from cerebral ischemia/reperfusion oxidative stress injury by directly targeting NOX2. Biomed Pharmacother. 2020, 124, 109860. [Google Scholar] [CrossRef] [PubMed]
- Zhu, M.L.; Zhang, J.; Guo, L.J.; et al. Amorphous selenium inhibits oxidative stress injury of neurons in vascular dementia rats by activating NMDAR pathway. Eur J Pharmacol. 2023, 955, 175874. [Google Scholar] [CrossRef] [PubMed]
- Parker, E.; Aboghazleh, R.; Mumby, G.; et al. Concussion susceptibility is mediated by spreading depolarization-induced neurovascular dysfunction. Brain. 2022, 145, 2049–2063. [Google Scholar] [CrossRef] [PubMed]
- El-Mezayen, N.S.; Abd, E.M.R.; El-Rewini, S.H. Vitamin B12 as a cholinergic system modulator and blood–brain barrier integrity restorer in Alzheimer's disease. Eur J Pharm Sci. 2022, 174, 106201. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



