Submitted:
03 July 2025
Posted:
04 July 2025
You are already at the latest version
Abstract
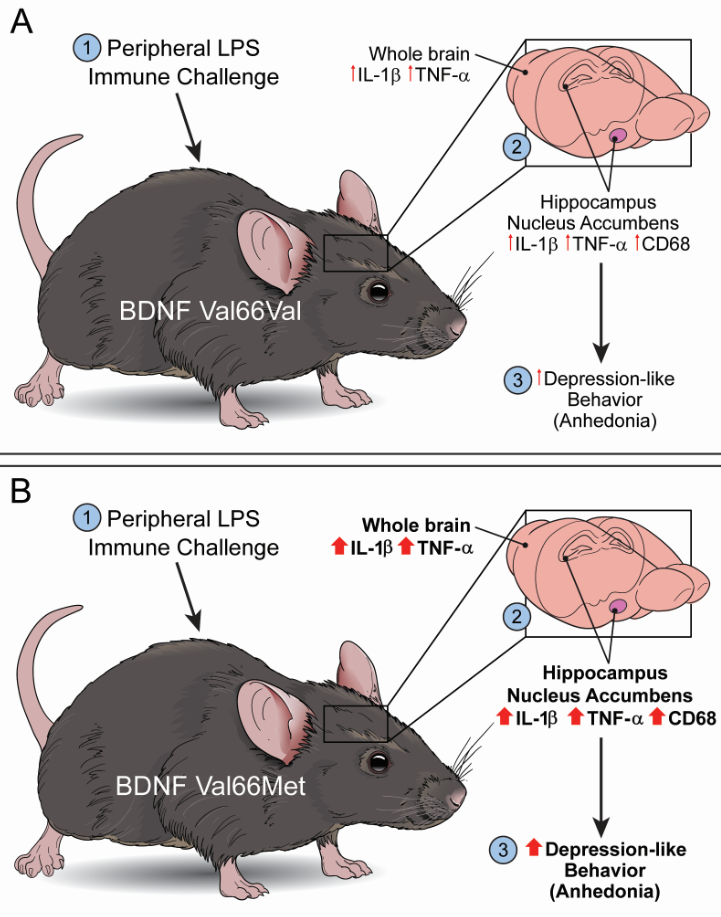
Dysregulated inflammatory processes contribute to depression, and gene-environment interactions may influence an individual’s risk and resilience. Reduced brain-derived neurotrophic factor (BDNF) expression increases susceptibility for developing depressive symptoms and the Val66Met (rs6265) single nucleotide polymorphism (SNP) on the BDNF gene is linked to mood disorders. However, whether Val66Met confers increased vulnerability to inflammation-induced depressive tendencies is unknown. Here, we tested the hypothesis - Val66Met SNP increases vulnerability to inflammation-induced depressive symptoms in a mouse model of lipopolysaccharide (LPS)-induced depression-like behavior. Behavior and neuroinflammation, following a 24hr LPS challenge, were measured in mice expressing human BDNF Val66Met gene variant or Val66Val littermates (control). The Val66Met genotype did not affect the peripheral inflammatory response, acute neuroinflammation, or the acute sickness behavior response. Val66Met mice exhibited anhedonia-like behavioral response following LPS challenge, and we found increased mRNA expression of IL-1β and TNFα in the cerebrum compared to controls. The mRNA expression of IL-1β and TNFα in the hippocampus and the nucleus accumbens of Val66Met mice were increased following LPS, and an interaction was detected for CD68 expression in the nucleus accumbens. In summary, these data suggest immune activation in Val66Met mice increased susceptibility to anhedonic behavior and dysregulated negative regulation of inflammation.
Keywords:
BDNF
; Neuroinflammation
; Depression
; LPS
Main Points:
Val66Met genotype did not affect peripheral inflammatory response, acute neuroinflammation, or acute sickness behavior response to Lipopolysaccharide
Val66Met genotype exacerbates LPS-induced anhedonia-like behavior and the neuroinflammatory response
BDNF Val66Met polymorphism may disrupt negative regulation of inflammation following peripheral immune challenge
1. Introduction
Depression is the leading cause of disability world-wide [1], and approximately 15-30% of patients experiencing depressive episodes respond inadequately to first line therapeutics [2]. Inflammation can induce depressive episodes [3] and increased inflammation can predict depressive symptoms [4]. Depressed patients with defective anti-inflammatory immune responses respond inadequately to antidepressant treatment [5], and blockade of inflammation (tumor necrosis factor antagonist – infliximab) had antidepressant efficacy in depressed patients with elevated C-reactive protein (CRP) levels [6]. Meta-analyses indicate that the inflammatory marker CRP is frequently elevated in individuals experiencing depression despite adjustment for epidemiological and other confounding factors. The increased CRP levels are associated with physical (appetite and energy) and cognitive symptoms of depression (lack of interest in activities); however, peripheral inflammation is not associated with emotional symptoms (hopelessness, feeling like a failure) of depression [7]. Thus, the variability in the severity of depressive symptoms experienced by individuals could be related to interactions between biological risk factors such as genetics and other environmental factors, such as inflammation or chronic stress. An understanding of the mechanisms underlying such interactions is needed to advance the tools required for personalized psychiatric and clinical care.
The neurotrophin hypothesis of depression postulates brain-derived neurotrophic factor (BDNF) to be involved in depression, and the implications of reduced/impaired BDNF are well established in human and animal studies [8]. Reduced serum levels of BDNF were observed in patients suffering major depressive disorder [9] and reduced brain gene-transcript and protein levels of BDNF were documented in young suicide victims [10]. Increased DNA methylation of the BDNF gene in peripheral blood cells of major depressive disorder patients suppresses BDNF expression to suggest that epigenetic changes to this neurotrophic gene may disrupt physiological BDNF-Tropomyosin receptor kinase B (TrkB) signaling [11]. Patients with depressive episodes exhibit reduced BDNF levels in the serum [12] and in the brain [13], while effective treatment with antidepressants increases BDNF levels in the brains of depressed patients [14]. In rodent models, chronic unpredictable stress induces depressive-like behaviors concomitant with reducing levels of mature BDNF and its receptor TrkB in the neocortex and the hippocampus [15]. However, the efficacy of an anti-depressant, like fluoxetine, to rescue depressive behaviors and increase BDNF mRNA is dependent on the environmental factors in which treatment is presented and outcomes vary between stressful and stress-free conditions [16]. Furthermore, previous investigations studying BDNF heterozygous mice suggest exposure to stress induces development of depressive-like behaviors [17,18] and genetic reduction of BDNF is associated with an exaggerated neuroimmune response to peripheral immune challenge [19]. While the relationship between the BDNF and neuroinflammation may be bidirectional in nature [20], the cellular and molecular mechanisms altered by such a gene (BDNF) and environment (immune challenge) interaction remain widely unknown.
A common single nucleotide polymorphism (SNP), rs6265, involving a valine for methionine substitution at amino acid position 66 (Val66Met) on the brain-derived neurotrophic factor (BDNF) gene is associated with development of dysthymic mood disorders [21], and the onset of cognitive [22] and memory deficits [23]. The Val66Met SNP impairs neuronal processing and reduces activity-dependent release of mature BDNF [23,24,25]. The BDNF Val66Met genotype is also linked to negative modulation of early life experiences, increasing the risk of developing depression in adulthood [26,27]. As such, the Val66Met SNP may be a vulnerability factor for stress-induced [28] or inflammation-induced depression [29]. Patients carrying the Val66Met allele were more likely to develop depressive symptoms after treatment with interferon-α [30], and the Met allele predicted inflammation-associated depressive symptoms in women with breast cancer [31]. In preclinical models, mice expressing the human BDNF Val66Met SNP under the endogenous BDNF promoter are more susceptible to anxiogenic stimuli, stress-induced depressive-like behaviors, and less responsive to antidepressant treatment [25,32]. These data suggest that the Val66Met SNP may sensitize the development of depressed mood associated with an aberrant inflammatory tone.
To directly investigate the interaction between the Val66Met SNP and neuroinflammation, we utilized mice expressing the human BDNF gene with or without the Val66Met SNP in the well-validated endotoxin (LPS)-induced depressive-like behavioral paradigm. In this study, we tested the hypothesis that the presence of the Val66Met SNP increases vulnerability to inflammation-induced depressive-like behaviors. Peripheral LPS challenge-induced changes were investigated using validated behavioral assays and peripheral inflammatory response by measuring changes in circulating cytokine levels, and measurement of inflammatory factors in the whole brain and in the hippocampus and in the nucleus accumbens. In this study, we aimed to provide the first insight into how Val66Met confers vulnerability to inflammation-induced depressive symptoms.
2. Methods and Materials
2.1. Animals
All animal care and use were carried out in accordance with the Guide for the Care and Use of Laboratory Animals, 8th edition (NRC) and approved by the Institutional Animal Care and Use Committees at the Audie L. Murphy VA Hospital and UT Health San Antonio. Male 10-16-week-old heterozygous BDNF Val66Met mice and wild-type Val66Val littermate controls on a C57BL/6J background were utilized in all experiments. The breeding colony was maintained by crossing heterozygous Val66Met male mice with wild-type Val66Val females. Pilot experiments identified differences in baseline behavioral phenotype of female mice, which precluded the combination of male and female mice in the same groups and funding support was insufficient to include full analysis of female mice. Thus, all experiments in this study were conducted in young-adult male mice. Mice were group-housed in standard shoebox cages within a ventilated caging system. Mice were allowed ad libidum access to food and water and maintained under a reverse 12:12 h light/dark with lights out at 11:00am. All animals used in this study were weighed and handled daily for 5 mins per mouse beginning three days prior to injections and subsequent behavioral testing. This handling helps to minimize stress and habituate mice to the experimenter. Animals were moved from the vivarium to the testing room 1-hour before testing began to acclimatize to the environment where testing was conducted. Cages were covered with an opaque curtain during transport minimize disruptions in circadian rhythms. All behavioral testing was conducted during the first four hours of the dark phase under low light conditions (<100 lux) to minimize stress associated with light, and animals were returned to home-cages and kept separated from animals undergoing testing.
2.2. Genotyping
DNA from experimental mice was used for genotyping using the following protocol: 8.5 µl JumpStart Taq (Sigma-Aldrich, St. Louis, MO), 0.85 µL primers (12 µM), 4.4 µL PCR-grade water, and 2 µL DNA. Primers sequences (Integrated DNA Technologies, Coralville, IA): common forward TCA TAC TTC GGT TGC ATG AAG G, Val reverse ATC CAG CAG CTC TTC GAT GAC G, and Met reverse ATA AAT CCA CTA GTG GTG GTG G. Upon receipt of the primers, they were reconstituted using PCR grade water to 100 µM, and aliquots were stored at -20°C. Primers remained stable for about 4 months. PCR was performed in a thermocycler (C1000 Bio-Rad, Hercules, CA): step 1) 94 °C, 3 min, step 2) 94 °C 1 min, step 3) 55 °C, 1 min, step 4) 72°C, 1 min, step 5) repeat steps 2-4 39x, step 5) 72°C, 5 min, step 6) hold at 10 °C. Loading dye was added to samples, then run at 75 V for 30 min through a 2.5% agarose gel, and DNA bands were visualized using SyberSafe reagent (ThermoFisher, Waltham, MA) and UV light.
2.3. Experimental Timeline and Treatments
Young Val66Val and Val66Met male mice (10-16 weeks old) on a C57BL/6J background were injected intraperitoneally (ip.) with 0.83 mg/kg lipopolysaccharide (LPS, E. coli 0127: B8, Sigma-Aldrich) or vehicle (0.9% sterile saline, Bound Tree Medical, Dublin, OH) as illustrated in Figure 1. Two or 24 hours after treatment, separate groups of mice were subjected to behavioral testing as described below. For molecular and cellular analyses, mice were euthanized at the indicated times after treatment, blood samples were collected from the inferior vena cava, and the mice were perfused transcardially with ice-cold heparinized saline. The whole brains were removed, and tissue was prepared for subsequent analysis.
2.4. Behavior Testing
Sucrose Preference:Anhedonia-like behavior was measured as previously described [33]. Briefly, mice (n=10 mice/group) were trained using a two-bottle free choice paradigm (1% sucrose or water) for three days prior to treatment. Sucrose preference was calculated as the 24 hours change in weight of sucrose / (weight of sucrose + water) * 100 following LPS challenge.
Open Field: To evaluate locomotor activity and anxiety-like behavior, the open field test was used as previously described [33]. Briefly, mice (n=5-7 mice/group) were placed in an open field arena for 5 minutes and their locomotive activity was video recorded. Videos were analyzed using Noldus software (Ethovision 8.5, Leesburg, VA).
Forced Swim Test: Behavioral despair-like behavior was measured as previously described [34]. Briefly, mice (n=6-7 mice/group) were placed in a 4L Nalgene beaker of water (25º C ± 1) for 5 minutes and their activity was video recorded from above. A trained observer blinded to experimental treatments then scored the videos by measuring the time the mouse spent immobile.
Social Exploration: To measure acute sickness-like behavior, social interaction of a novel conspecific was measured as previously described [35] with minor modifications. Briefly, after habituation to an open field arena with an empty inverted mesh cup in the corner for 15 minutes, a novel same-sex conspecific was placed under the cup and activity of the test mouse was video recorded for 10 mins (n=9-11 mice/group). Videos were analyzed using Noldus software (Ethovision 8.5).
Y-Maze: Short-term working memory was assessed using a two-trial Y-maze task [36]. Distinct visual intra-maze cues [37] were posted at the end of each arm during a 15-minute exposure trial in which one of the arms was blocked. After 1 hour, the mouse was reintroduced to the same maze with free access to all three arms, and video-recorded for 5 minutes. Videos were analyzed using the Noldus software (Ethovision 8.5). Discrimination ratio was calculated as (time spent in novel arm) / (time spent in novel + familiar arms) (n=8-10 mice/group).
2.5. Tissue Preparation and RT-qPCR
Saline perfused brain tissue was flash frozen in liquid nitrogen and stored at -80 °C until homogenization in RNA lysis buffer from the PureLink RNA mini kit (ThermoFisher) using a BeadRuptor (Omni International, Kennesaw, GA). Samples were then centrifuged at 4°C for 10 mins at 1500 x g to pellet the beads and insoluble tissue pieces. The supernatant was removed for RNA isolation according to manufacturer instructions. Brain hemispheres were micro-dissected to collect the hippocampus and nucleus accumbens according to stereotaxic coordinates in the Franklin and Paxinos mouse brain atlas [38], then processed as described above.
RNA concentrations were determined using a spectrophotometer (NanoVue Plus, GE Healthcare, Chicago, IL), then equal concentrations of RNA were used for reverse transcription to generate cDNA using the High-Capacity cDNA reverse transcription kit (Applied Biosystems, Waltham, MA). cDNA was used in RT-qPCR using Taqman primers and probes (ThermoFisher; Gapdh Mm99999915, Il1b Mm00434228_m1, Tnfa Mm00443258, Il6 Mm00446190, Aif1 Mm00479862, Cd68 Mm03047343_m1, Nos2 Mm00440502, IL4Ra Mm01275139_m1, Il1Rn Mm00446186_m1). RT-qPCR was performed using the Bio-Rad CFX 384 system. Data are expressed as fold change relative to Val66Val saline controls using the ΔΔCt method [39].
2.6. Enzyme-Linked Immunosorbent Assay
Plasma concentrations of the pro-inflammatory cytokines - interleukin-1β (IL-1β), interleukin-6 (IL-6), and tumor necrosis factor-α (TNFα) were determined using ELISA kits (Invitrogen, Waltham, MA) according to manufacturer instructions.
2.7. Statistical Analysis
Data were checked to ensure that normality of distribution assumptions were met using the Shapiro-Wilk test. All data were analyzed using GraphPad Prism10.4.1 (San Diego, CA) using two-way ANOVA with genotype and LPS challenge as the two factors when assumptions of normality were met. If there was a significant interaction between the factors, Tukey’s multiple comparison post hoc test was used to determine significant differences between means of the different treatment groups. Non-normally distributed data were analyzed using the non-parametric Kruskal Wallis’s test and for post-hoc testing, the corrected Dunn’s multiple comparison test was used to determine significant difference between means of the different genotype and treatment groups. Data were considered significant if p<0.05. Spurious data were identified using Chauvenet’s outlier criteria [40] and a total of 5 animals were excluded from data analysis.
3. Results
3.1. BDNF Val66Met Expression Results in Susceptibility to the Anhedonia-Like Behavioral Effects of Immune Challenge
Following an intraperitoneal LPS challenge, Val66Met mice did not exhibit a preference for sucrose solution over water, and their sucrose preference was significantly lower than saline-treated same genotype controls or Val66Val mice challenged with LPS (Genotype x LPS interaction: F (1,36) = 6.285; p=0.0168, η2p = 0.149; Figure 1A). The duration of immobility (non-normal distribution) in the forced swim test (FST) was not different between saline-treated controls, and LPS challenge did not result in differences in the FST between Val66Val and Val66Met mice (Kruskal-Wallis’s test, p=0.6397; Figure 1B). The effect of immune activation (LPS: F (1,20) = 6.088, p=0.0228, η2p = 0.407) on thigmotaxic (wall-hugging) behavior persisted in both genotypes in the open field test (Figure 1C), and group means were not different from each other (Tukey’s multiple comparison testing). There were no significant differences in locomotor activity in the open field test 24 hours after LPS challenge. In the trial phase of the Y-maze, there were no baseline differences in performance related to genotype and LPS challenge did not affect ability to discriminate between the novel (blocked) versus the familiar (unblocked) (Figure 1D).
Two hours after LPS, mice of both genotypes exhibited sickness-like behavior, with a main effect of LPS dampening locomotion (LPS: F (1,20) = 33.81, p<0.0001, η2p = 0.628) and social exploration (Kruskal-Wallis’s test H(3) = 10.24, p=0.0166; Supplementary Figure S1). 24 hours after immune challenge (LPS: F (1,56) = 146.2, p<0.0001, η2p = 0.723), both genotypes lost similar amounts of body weight (Figure 1E), and post hoc testing revealed significant differences between saline and LPS treated Val66Val mice (p<0.0001) and Val66Met mice (p<0.0001). 24 hours after immune challenge, no significant changes in locomotor activity or social exploration were observed (Figure 1F). Finally, at 2 and 24 hours post-LPS challenge, no significant interactions between treatment and genotype were detected in plasma concentrations of the pro-inflammatory cytokines IL-1β, IL-6, or TNFα (Table 1).
3.2. Immune Challenge Induces Dysregulated Whole Brain Pro-Inflammatory Cytokine Expression in BDNF Val66Met Mice
At 2, or 24 hours after peripheral LPS challenge, brain cerebral hemispheres were collected to analyze mRNA expression using real time RT-PCR (Figure 2). At 2 hours, LPS-induced immune challenge increased the expression of the pro-inflammatory cytokines IL-1β (LPS: F (1,10) = 36.28; p=0.0001, η2p = 0.784) and TNFα (Kruskal-Walli’s test H (3) = 12.63, p=0.0055, η2 = 0.574) (Figure 2A,B). Tukey’s multiple comparison post hoc testing found LPS treated Val66Val (p=0.0146) and Val66Met (p=0.0035) mice were different from saline treated controls on the mRNA expression of IL-1β. The mRNA expression of TNFα was increased in LPS challenged Val66Val mice (Dunn’s multiple comparison test, p=0.0372) compared to saline controls while the expression of TNFα was not different in brain tissue of saline and LPS treated Val66Met mice.
At 24 hours after immune challenge when the LPS induced sickness behavior is resolved, (Supp. Figure 1), we detected a significantly higher expression of the pro-inflammatory cytokines IL-1β (Kruskal-Wallis’s test H(3) = 19.85, p=0.0002, η2 = 0.735) and TNFα (Genotype x LPS: F (1,21) = 4.512, p=0.0457, η2p = 0.177) in the brain tissue of LPS challenged Val66Met mice in comparison to saline treated controls and Val66Val mice (Figure 2C,D). At 24 hours, for changes in mRNA expression of IL-1β, Dunn’s multiple comparison test found significant difference between saline and LPS-treated Val66Met mice (p=0.0004). There were no significant interactions in expression of several other pro- and anti-inflammatory factors (Supplementary Table S1).
3.3. BDNF Val66Met Mutation Differentially Affects Brain Regions in Response to LPS
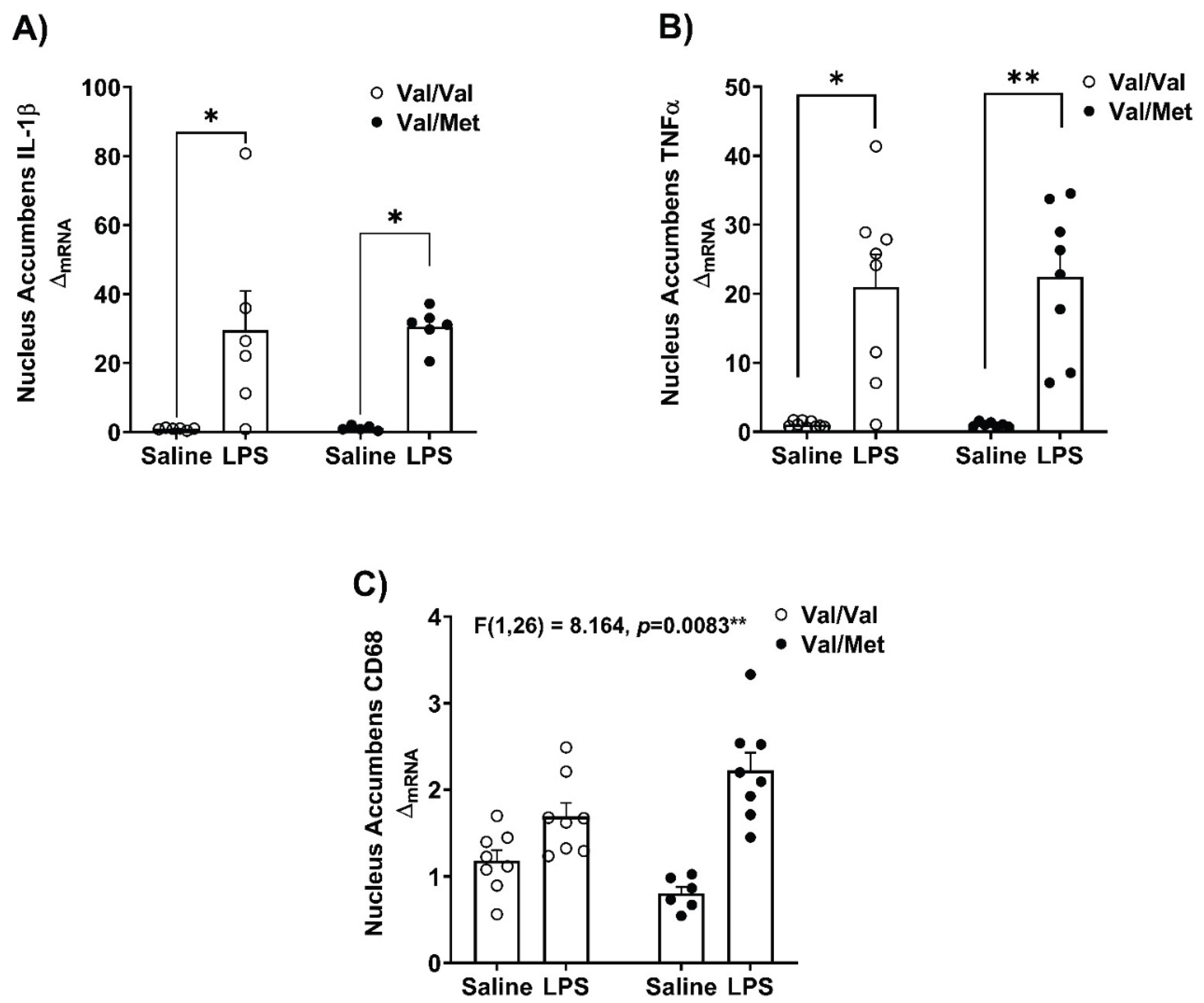
Steady state mRNA expression of inflammatory cytokines and lysosomal/phagocytic marker - CD68, were measured in the hippocampus and the nucleus accumbens (Figure 3 and Figure 4). We investigated these brain regions as they are involved in the regulation of behaviors affected in depression [41,42,43,44]. We found increased mRNA expression of IL-1β (Kruskal-Wallis’s test H (3) = 12.63, p=0.0055, η2 = 0.486) and TNFα (Kruskal-Wallis’s test H (3) = 20.55, p=0.0001, η2 = 0.735) in the hippocampus of Val66Met mice (Figure 3A,B). Similarly, in the nucleus accumbens of Val66Met mice, (Figure 4A,B) LPS challenge influenced the mRNA expression of IL-1β (Kruskal-Wallis’s test H (3) = 15.53, p=0.0014, η2 = 0.597) and TNFα (Kruskal-Wallis’s test H (3) = 20.98, p=0.0001, η2 = 0.677). Exploratory post-hoc testing found no differences of the expression of these gene transcripts between LPS-treated Val66Val and Val66Met mice.
In the hippocampus, mRNA expression of CD68 was higher in LPS-treated Val66Met mice (Kruskal-Walli’s test H (3) = 16.22, p=0.0010, η2 = 0.559) but not in saline or LPS Val66Val mice (Figure 3C). In the nucleus accumbens, we found an interaction between genotype and LPS challenge on the mRNA expression of CD68 (Genotype x LPS F (1,26) = 8.164, p=0.0083, η2p = 0.239) (Figure 4C).
Figure 5.
Measurement of pro-inflammatory factors in the Nucleus Accumbens in Val66Val and Val66Met mice in response to peripheral immune activation. After 24 hours, whole brains were micro-dissected and PCR was performed to determine relative gene expression in the hippocampus, and data were analyzed using a two-way ANOVA with genotype and LPS as independent factors for normally distributed data and non-parametric Kruskal-Wallis’s test were used to analyze non-normally distributed data. The mRNA expression of A) IL-1β, Kruskal-Wallis’s test H (3) = 15.53, p=0.0014, η2 = 0.597; B) TNFα, Kruskal-Wallis’s test H (3) = 20.98, p=0.0001, η2 = 0.677). C) CD68, genotype x LPS interaction: F (1,26) = 8.164, p=0.0083, η2p = 0.239. n=4-8 mice/group; Post-hoc testing with Tukey’s multiple comparison test to compare differences between group means if main effects of genotype or LPS were detected and for non-normally distributed data post-hoc testing with corrected Dunn’s multiple comparison test was used to compare differences between group means; *p<0.05, **p<0.01, ***p<0.001.
Figure 5.
Measurement of pro-inflammatory factors in the Nucleus Accumbens in Val66Val and Val66Met mice in response to peripheral immune activation. After 24 hours, whole brains were micro-dissected and PCR was performed to determine relative gene expression in the hippocampus, and data were analyzed using a two-way ANOVA with genotype and LPS as independent factors for normally distributed data and non-parametric Kruskal-Wallis’s test were used to analyze non-normally distributed data. The mRNA expression of A) IL-1β, Kruskal-Wallis’s test H (3) = 15.53, p=0.0014, η2 = 0.597; B) TNFα, Kruskal-Wallis’s test H (3) = 20.98, p=0.0001, η2 = 0.677). C) CD68, genotype x LPS interaction: F (1,26) = 8.164, p=0.0083, η2p = 0.239. n=4-8 mice/group; Post-hoc testing with Tukey’s multiple comparison test to compare differences between group means if main effects of genotype or LPS were detected and for non-normally distributed data post-hoc testing with corrected Dunn’s multiple comparison test was used to compare differences between group means; *p<0.05, **p<0.01, ***p<0.001.
4. Discussion
In this study, we used the mouse model of LPS-induced neuroinflammation to test the hypothesis that expression of the Val66Met BDNF SNP increases vulnerability to inflammation-induced depressive-like behaviors. The data suggests that mice expressing the human BDNF gene with Val66Met SNP exhibited pronounced anhedonia-like behavior following immune challenge. Thigmotactic behavior was higher following LPS challenge in both Val66Val and Val66Met genotypes, but LPS challenge did not affect short-term working memory or sociability in either genotype. The cerebral mRNA expression of inflammatory cytokines (IL-1β, TNFα) was higher in Val66Met mice compared to Val66Val mice at 24hrs after LPS challenge whereas cerebral mRNA expression of the microglia marker, IBA1 (data not shown), was higher in both LPS treated Val66Val and Val66Met mice. Further, LPS challenge increased the expression of IL-1β and TNFα in the hippocampus and in the nucleus accumbens. In these same brain regions, the mRNA expression of CD68 was significantly higher in LPS challenged Val66Met mice compared to saline-treated genotype controls. Collectively, these results suggest that following an immune challenge, Val66Met mice have higher susceptibility for development of anhedonia-like behavior and have increased cerebral neuroinflammation.
The role of BDNF in the etiology of depression remains widely debated [45,46], however, evidence suggests BDNF signaling is involved in the effectiveness of anti-depressant drugs (14, 32, 47). The Val66Met SNP may increase sensitivity to stress-induced depression [47,48]; meta-analysis have found Val66Met polymorphism in the BDNF gene moderates the relationship between stress and depression [28]. Genetic factors that interact with external stressors, including stress and inflammation could be involved in increasing risk for mood dysregulation [19,20]. Inflammation represents an important risk factor, but little is known regarding genetic contributions that modulate risk for developing depressive behaviors in response to inflammation. The BDNF Val66Met polymorphism [47,49] and LPS-induced immune activation [33,50] have been independently linked to the development of depressive and anxiety-like behaviors. To date, two clinical studies have linked BDNF Val66Met polymorphism with inflammation-associated depressive symptoms [29,31]. However, whether the Val66Met SNP directly interacts with neuroinflammatory processes to potentiate the depressive-like behavioral effects of inflammation, has not been investigated. Here, we found LPS challenge in Val66Met mice led to decrease in sucrose preference suggesting anhedonia-like response independent of sickness-like behavior. LPS induced neuroinflammation has been shown to increase dopamine synthesis and release in the nucleus accumbens [51], that suggests that these dopamine deficits specifically impair motivational drive required for reward-seeking behavior without interference from malaise or fatigue caused by LPS but rather are related to specific effect of increased pro-inflammatory cytokine production following immune challenge [3,52,53,54]. Further, we did not find reductions in motivations to complete other behavioral tests like FST, Y-maze or social exploration suggesting that at 24 hours after LPS challenge, acute sickness behavior was resolved and did not interfere or influence behavioral testing which is consistent with our line of inquiry [55]. However, the increased thigmotactic behavior induced by LPS was not affected/influenced by the Val66Met polymorphism. We did not find differences related to Val66Met genotype and LPS challenge on short-term working memory or in social exploratory behavior. Interestingly, our prior study using BDNF heterozygous mice revealed a similar depressive-like phenotypic response to LPS featuring a reduction in sucrose preference without concomitant neurovegetative-like behavioral potentiation [19]. These data begin to suggest that, perhaps, BDNF activity plays an important role in regulating anhedonia-like behavior during inflammatory conditions.
Recent computational analysis suggests a bi-directional relationship between systemic inflammatory regulators and the development of psychiatric disorders [56]. Peripheral inflammatory signals are communicated to the central nervous system by various routes [55,57,58], including the neural (Vagus nerve)-dependent route and the humoral route via secreted cytokines acting at the blood brain barrier as well across it [51]. In response, infiltrating and brain resident immune cells trigger/orchestrate a neuroinflammatory response characterized by increased production of inflammatory cytokines [59,60,61,62]. Using the well-validated LPS induced immune activation model, we found increased cerebral mRNA expression of IL-1β and TNFα in both Val66Val and Val66Met mice, but the magnitude was significantly higher in the brain tissue of Val66Met mice. This indicates heightened sensitivity in brain tissue to immune activation in mice carrying the Val66Met polymorphism. In the hippocampus and in the nucleus accumbens mRNA expression of IL-1β and TNFα was higher following LPS induced immune activation in the Val66Met genotype. It is plausible that infiltrating monocytes and macrophages could contribute to differential neuroinflammatory response in the brain, like previously published reports [63,64,65], however, additional studies are necessary to confirm this speculation in the context of the Val66Met polymorphism. In the nucleus accumbens, the mRNA expression of CD68 was modulated by both immune activation and the Val66Met polymorphism. CD68 is a known marker of microglia and macrophages during inflammatory states and critically involved in lysosomal/phagocytic activity of these cells [66,67,68]. Increased CD68 expression has been linked to stress and inflammation-induced depressive behaviors [69,70,71]. One study found mice expressing the Val66Met polymorphism to have higher CD68 on myocardial macrophages [72]. To our knowledge, this is the first study to investigate CD68 mRNA expression within context of neuroinflammation and the BDNF Val66Met polymorphism - we found that CD68 transcripts were modulated by Val66Met polymorphism and LPS challenge and suggests a differential neuroinflammatory response in the nucleus accumbens. We focused and limited our investigations of inflammatory response in the nucleus accumbens and the hippocampus, as several reports have found links between the BDNF Val66Met polymorphism differentially affecting theses brain regions to modulate anxiety-like and depressive-like behaviors [25,47,48,73,74]. Neuroinflammation triggered by injecting LPS directly into the nucleus accumbens of mice is related to growth factors (progranulin) and NF-κB signaling pathway which are closely associated with innate immune response and produce a similar behavioral and neuroinflammatory profile we report in this study [75,76], and this study provides further evidence for a role of the BDNF Val66Met polymorphism in this context. Within the nucleus accumbens, LPS triggered neuroinflammation and development of depressive-like behaviors are linked to activity at the dopamine D3 receptor that are directly associated with motivational and reward-related behaviors like anhedonia, again like observations documented here in LPS treated BDNF Val66Met mice [77]. In contrast, in the hippocampus, we observed increased cytokine transcripts and CD68 only in Val66Met mice injected with LPS indicating the potential sensitivity of the hippocampus for gene x environment interactions in carriers of the Val66Met SNP. Importantly, the hippocampus is a critical brain region for learning and memory and while it has been implicated in both anxiety and depressive disorders, our results suggest that anhedonia-like behavior as well as neuroinflammatory response are closely related to the interaction between BDNF Val66Met polymorphism and LPS challenge in the nucleus accumbens. Early studies using these Val66Met mice reported extensively on their sensitivity to stress-based challenges or fear-based paradigms. To our knowledge, this study is the first to report, albeit with a limited focus, the functional neuroinflammatory and behavioral consequences of the Val66Met SNP in the LPS-induced depressive-like behavioral paradigm. This study did not include behavioral testing targeting the hippocampus such as Barnes maze or learned helplessness which would help reveal changes in spatial learning and depressive symptoms respectively [78]. However, it is critical to note that the BDNF Val66Met polymorphism also affects the effectiveness of antidepressants [79,80], especially in the hippocampus that appears highly sensitive to the Val66Met polymorphism following stress exposure [81,82,83]. However, the present results are consistent with our previous data reporting a more profound anhedonia-like response to LPS in BDNF +/- mice [19], which suggests potential specificity in the response that involved nucleus accumbens. Future studies are required to dissect stress v/s immune challenge associations with depressive-like behaviors and relevant changes in inflammatory markers.
Inflammation is increased by myriad factors, and in experimental settings, activation of the peripheral immune response leads to depressive symptoms in both humans and rodents [51,55,84,85]. The association between inflammatory cytokines and depression has been well described, with alterations in serum and plasma levels of IL-1β, IL-6, IFNγ and C-reactive protein (CRP) as common indicators of perturbed inflammatory tone and associated depressive symptoms [53,86]. In response to peripheral inflammatory mediators, the resident immune cells of the CNS – microglia, in turn, increase the upregulation and secretion of proinflammatory cytokines and chemokines. Among these, IL-1β and TNFα are important regulators of the acute sickness response, and consistent with the behavioral response observed in Supplementary Figure S1, plasma cytokines and mRNA transcripts of IL-1β and TNFα were increased in both Val66Val and Val66Met mice compared to controls at 2 hours following LPS challenge. However, behavioral differences apparent in Val66Met mice at 24 hours, were paralleled by a significantly higher expression of inflammatory gene transcripts in the brain in Val66Met mice (Figure 2). These data suggest that resolution of inflammation was impaired in Val66Met mice and could contribute to anhedonia-like behavior. Of note, no genotype x LPS interaction was apparent in peripheral cytokine protein levels that suggests the Val66Met SNP does not impact peripheral immune regulation, rather dysregulates neuroinflammation. These are the first data to reveal the specific effect of the BDNF Val66Met SNP on neuroinflammation. The results of this study build on our previous investigations where we have reported that BDNF heterozygous mice have increased vulnerability to develop stress induced anhedonia [18] and genetic reduction of BDNF was associated with reduced sucrose preference and increased neuroimmune response to peripheral immune challenge [19]. Importantly, inflammation alone does not explain the full extent of depressive symptoms [87] and, not all individuals develop depressive symptoms even in the face of increased inflammation. It is plausible that a better understanding of the interaction between inflammation and genetic/environmental factors documented in this study will help resolve these gaps in knowledge.
This study has several strengths and limitations. We have found that BDNF Val66Met mice exhibit an anhedonia-like behavioral response after LPS, while Val66Val mice did not (Figure 1). We ruled out the possibility of sickness behavior as a confounding factor as evidenced by data in Suppl. Figure 1. Acute and extended sickness-related behaviors did not change across genotypes, suggesting trajectory of sickness did not affect behavioral testing. No differences in exploration capacity, body weight, or locomotion (not shown) related to Val66Met SNP were observed, indicating absence of a general lethargy effect that confounded behavioral testing. However, a more detailed time-course study warrants future studies to investigate this further. We did not find behavioral differences in the FST, but other studies have found increased immobility using this measure in Val66Met mice under stressed conditions [47,49]. However, the internal validity of FST in measuring depressive-like behaviors in rodent models is not robust with variable results reported in the literature, and the FST has better sensitivity in assessing the effectiveness of antidepressant and anti-anxiety medications [88,89,90]. LPS induced immune activation increased thigmotactic (wall-hugging) behavior, in both genotypes. However, Val66Val mice were resilient to LPS challenge and did not exhibit anhedonia-like behavior (Figure 1A). The study by Notaras and colleagues found the Val66Val genotype to be resilient against mood maladaptation and required chronic corticosteroid administration to induce susceptibility to depressive behaviors [49]. Chen and colleagues observed an anxiety-like behavioral phenotype in BDNF Val66Met mice in the elevated plus maze; perhaps a stressor stronger than that of LPS like chronic corticosterone treatment is required to induce an anxiogenic phenotype [25,49]. Interestingly, humans expressing BDNF Val66Met show no baseline differences in several metrics of anxiety when compared to Val66Val subjects [91], and anxiety scores measured following pro-inflammatory treatment are comparable between Val66Val and Val66Met-expressing individuals [30].
Previous studies have noted significant sex-based differences in the behavioral and transcriptomic elements between male and female humanized BDNF knock-in mice [92]. Of note, female BDNF Val66Met mice exhibit an anxiogenic phenotype at baseline in an age and estrus cycle dependent manner [93]. The results presented here were performed in young-adult male humanized BDNF knock-in mice and limits the ability to comment on sex-based differences during an ongoing immune challenge. Regardless of limitations, the behavioral testing performed in male BDNF Val66Met mice did not reveal baseline performance deficits in sucrose preference or FST (Figure 1A,B). Future studies are required to investigate sex-based differences on neuroinflammation outcomes related to BDNF Val66Met SNP. Specifically, future research should include testing of female mice at different ages as well as ovariectomized (with and without) female Val66Val and Val66Met mice under immune challenge conditions to address sex-based differences. The observed differences in behavioral and neuroinflammatory response reported in this study suggest that LPS induced anhedonia-like behavior in Val66Met mice are only partially explained by localized changes in inflammatory gene transcripts measured in the hippocampus and the nucleus accumbens (Figure 3 and Figure 4). Interestingly, CD68 mRNA expression was remarkably different and suggests a role of microglia and/or infiltrating monocytes/macrophages in dysregulated inflammatory process but additional studies are needed to confirm this. Noteworthy, inflammatory markers studied in whole brain tissue (Figure 2) clearly indicate an upregulated expression in Val66Met mice treated with LPS. Speculatively, other brain regions such as the prefrontal cortex and the amygdala may be involved in mediating the neuroinflammatory response post LPS challenge. While prior studies have implied compromised BBB integrity as a contributing factor in neuroinflammatory responses to peripheral immune challenge, our unpublished data in the C57B6/J background strain did not identify any BBB breakdown at the dose of LPS being used in this study. If the Val66Met SNP rendered the BBB more vulnerable to LPS-induced breakdown, it would likely impact the BBB throughout the brain, but these important questions remain to be directly investigated.
BDNF facilitates neuronal survival, differentiation, and proliferation and plays an important role in neurogenesis and plasticity [94]. The Val66Met SNP is the most extensively studied BDNF SNP, and it may be a genetic risk factor contributing to depression by interacting with stress or inflammation. Building on previous reports where BDNF deficient mice exhibit protracted neuroinflammatory and behavioral responses following peripheral immune challenge with LPS [19,20], here, we provide evidence for BDNF Val66Met male mice to exhibit prolonged anhedonia-like behavioral effects of LPS, suggestive of a good model to dissect the mechanism(s) of differential depression vulnerability. Overall, this study revealed distinct SNP x LPS interaction that prolonged elements of the neuroinflammatory response, while no differences were observed in the peripheral inflammatory response or the acute sickness-related behavioral response. In conclusion, these data support that the BDNF Val66Met polymorphism may be an important contributor to increased vulnerability in the development of inflammation associated anhedonia-like behavior. The sustained elevation of IL-1β and TNFα expression specifically in the brain without significant differences in peripheral cytokine responses—This suggests that genetic vulnerability factors such as Val66Met may impair the resolution of neuroinflammation independently of peripheral inflammatory resolution. This may be particularly relevant when considering the potential implications of chronic or cumulative exposure to poorly resolved/regulated neuroinflammatory responses across the lifespan. These findings could have implications for individuals with the Val66Met polymorphism who might benefit from therapeutic strategies targeting central (rather than solely peripheral) inflammatory processes. Our results advocate additional studies that investigate personalized treatment approaches in depression, where inflammatory biomarkers and genetic screening for BDNF polymorphisms could inform the use of anti-inflammatory or neurotrophic-based interventions (e.g., TNF antagonists, IL-1 blockers, or agents promoting BDNF signaling such as TrkB agonists).
Funding Statement and Acknowledgements:
This research was supported by VA Merit Award I01BX003195-01, NIH National Centers for Advancing Translational Sciences grant ML1 TR001120 (JCO), NIH Institutional training grant T32 NS 082145 (AMG), Wilbur D. Mills Distinguished Chair in Alcoholism and Drug Abuse Prevention (AA). The content is the sole responsibility of the authors and does not necessarily represent the views of the National Institute of Mental Health or the National Institutes of Health. We would like to thank Dr. Francis Lee from Cornell University for kindly providing our founder BDNF Val66Met breeding mice.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org.
Author Contributions
Conceptualization, methodology, data curation, formal analysis, investigation, validation and visualization – AG, MM, MD, JCO. Funding acquisition, project administration, resources, supervision – JCO and AA. Writing – original draft – AG and MM. Writing – reviewing & editing – AG, MM, MD, SS, AA and JCO.
Data Availability Statement
Individual data points are provided in the figures and raw data used to generate the summary findings of this study are available from the corresponding author – Jason C. O’Connor, PhD – oconnorj@uthscsa.edu upon request.
Conflicts of Interest
All authors have no competing interests to disclose.
References
- Friedrich, M.J. Depression Is the Leading Cause of Disability Around the World. JAMA. 1517. [Google Scholar]
- Dodd, S.; Bauer, M.; Carvalho, A.F.; Eyre, H.; Fava, M.; Kasper, S.; Kennedy, S.H.; Khoo, J.P.; Lopez Jaramillo, C.; Malhi, G.S.; et al. A clinical approach to treatment resistance in depressed patients: What to do when the usual treatments don't work well enough? World J Biol Psychiatry. 2021, 22, 483–494. [Google Scholar] [CrossRef]
- Felger, J.C.; Haroon, E.; Miller, A.H. Risk and Resilience: Animal Models Shed Light on the Pivotal Role of Inflammation in Individual Differences in Stress-Induced Depression. Biological Psychiatry. 2015, 78, 7–9. [Google Scholar] [CrossRef]
- Gimeno, D.; Kivimäki, M.; Brunner, E.J.; Elovainio, M.; De Vogli, R.; Steptoe, A.; Kumari, M.; Lowe, G.D.O.; Rumley, A.; Marmot, M.G.; et al. Associations of C-reactive protein and interleukin-6 with cognitive symptoms of depression: 12-year follow-up of the Whitehall II study. Psychological Medicine. 2009, 39, 413–423. [Google Scholar] [CrossRef]
- Syed, S.A.; Beurel, E.; Loewenstein, D.A.; Lowell, J.A.; Craighead, W.E.; Dunlop, B.W.; Mayberg, H.S.; Dhabhar, F.; Dietrich, W.D.; Keane, R.W.; et al. Defective Inflammatory Pathways in Never-Treated Depressed Patients Are Associated with Poor Treatment Response. Neuron. 2018, 99, 914–924.e913. [Google Scholar] [CrossRef] [PubMed]
- Raison, C.L.; Rutherford, R.E.; Woolwine, B.J.; Shuo, C.; Schettler, P.; Drake, D.F.; Haroon, E.; Miller, A.H. A Randomized Controlled Trial of the Tumor Necrosis Factor Antagonist Infliximab for Treatment-Resistant Depression. JAMA Psychiatry. 2013, 70, 31–31. [Google Scholar] [CrossRef] [PubMed]
- Frank, P.; Jokela, M.; Batty, G.D.; Cadar, D.; Steptoe, A.; Kivimaki, M. Association Between Systemic Inflammation and Individual Symptoms of Depression: A Pooled Analysis of 15 Population-Based Cohort Studies. Am. J. Psychiatry. 2021, 178, 1107–1118. [Google Scholar] [CrossRef] [PubMed]
- Lee, B.-H.; Kim, Y.-K. The Roles of BDNF in the Pathophysiology of Major Depression and in Antidepressant Treatment. Psychiatry Investigation. 2010, 7, 231–231. [Google Scholar] [CrossRef]
- Molendijk, M.L.; Bus, B.A.A.; Spinhoven, P.; Penninx, B.W.J.H.; Kenis, G.; Prickaerts, J.; Voshaar, R.C.O.; Elzinga, B.M. Serum levels of brain-derived neurotrophic factor in major depressive disorder: State–trait issues, clinical features and pharmacological treatment. Molecular Psychiatry. 2011, 16, 1088–1095. [Google Scholar] [CrossRef]
- Pandey, G.N.; Ren, X.; Rizavi, H.S.; Conley, R.R.; Roberts, R.C.; Dwivedi, Y. Brain-derived neurotrophic factor and tyrosine kinase B receptor signalling in post-mortem brain of teenage suicide victims. The International Journal of Neuropsychopharmacology. 2008, 11, 1047–1047. [Google Scholar] [CrossRef]
- Roy, B.; Shelton, R.C.; Dwivedi, Y. DNA methylation and expression of stress related genes in PBMC of MDD patients with and without serious suicidal ideation. J. Psychiatr. Res. 2017, 89, 115–124. [Google Scholar] [CrossRef]
- Bus, B.A.; Molendijk, M.L.; Tendolkar, I.; Penninx, B.W.; Prickaerts, J.; Elzinga, B.M.; Voshaar, R.C. Chronic depression is associated with a pronounced decrease in serum brain-derived neurotrophic factor over time. Mol. Psychiatry. 2015, 20, 602–608. [Google Scholar] [CrossRef]
- Duman, R.S.; Monteggia, L.M. A neurotrophic model for stress-related mood disorders. Biol. Psychiatry. 2006, 59, 1116–1127. [Google Scholar] [CrossRef] [PubMed]
- Castren, E.; Monteggia, L.M. Brain-Derived Neurotrophic Factor Signaling in Depression and Antidepressant Action. Biol. Psychiatry. 2021, 90, 128–136. [Google Scholar] [CrossRef] [PubMed]
- Bai, Y.Y.; Ruan, C.S.; Yang, C.R.; Li, J.Y.; Kang, Z.L.; Zhou, L.; Liu, D.; Zeng, Y.Q.; Wang, T.H.; Tian, C.F.; et al. ProBDNF Signaling Regulates Depression-Like Behaviors in Rodents under Chronic Stress. Neuropsychopharmacology. 2016, 41, 2882–2892. [Google Scholar] [CrossRef] [PubMed]
- Alboni, S.; van Dijk, R.M.; Poggini, S.; Milior, G.; Perrotta, M.; Drenth, T.; Brunello, N.; Wolfer, D.P.; Limatola, C.; Amrein, I.; et al. Fluoxetine effects on molecular, cellular and behavioral endophenotypes of depression are driven by the living environment. Mol. Psychiatry. 2017, 22, 552–561. [Google Scholar] [CrossRef]
- Duman, C.H.; Schlesinger, L.; Kodama, M.; Russell, D.S.; Duman, R.S. A Role for MAP Kinase Signaling in Behavioral Models of Depression and Antidepressant Treatment. Biological Psychiatry. 2007, 61, 661–670. [Google Scholar] [CrossRef]
- Dugan, A.M.; Parrott, J.M.; Redus, L.; Hensler, J.G.; O’Connor, J.C. Low-Level Stress Induces Production of Neuroprotective Factors in Wild-Type but Not BDNF <sup>+/-</sup> Mice: Interleukin-10 and Kynurenic Acid. International Journal of Neuropsychopharmacology. 2016, 19, pyv089–pyv089. [Google Scholar]
- Parrott, J.M.; Porter, G.A.; Redus, L.; O'Connor, J.C. Brain derived neurotrophic factor deficiency exacerbates inflammation-induced anhedonia in mice. Psychoneuroendocrinology. 2021, 134, 105404. [Google Scholar] [CrossRef]
- Porter, G.A.; O'Connor, J.C. Brain-derived neurotrophic factor and inflammation in depression: Pathogenic partners in crime? World J Psychiatry. 2022, 12, 77–97. [Google Scholar] [CrossRef]
- Lee, S.Y. The Correlation between Plasma Brain-derived Neurotrophic Factor and Cognitive Function in Bipolar Disorder is Modulated by the BDNF Val66Met Polymorphism. European Psychiatry. 2017, 41, S76–S76. [Google Scholar] [CrossRef]
- Soliman, F.; Glatt, C.E.; Bath, K.G.; Levita, L.; Jones, R.M.; Pattwell, S.S.; Jing, D.; Tottenham, N.; Amso, D.; Somerville, L.H.; et al. A Genetic Variant BDNF Polymorphism Alters Extinction Learning in Both Mouse and Human. Science. 2010, 327, 863–866. [Google Scholar] [CrossRef]
- Egan, M.F.; Kojima, M.; Callicott, J.H.; Goldberg, T.E.; Kolachana, B.S.; Bertolino, A.; Zaitsev, E.; Gold, B.; Goldman, D.; Dean, M.; et al. The BDNF val66met Polymorphism Affects Activity-Dependent Secretion of BDNF and Human Memory and Hippocampal Function. Cell. 2003, 112, 257–269. [Google Scholar] [CrossRef]
- Adachi, N. New insight in expression, transport, and secretion of brain-derived neurotrophic factor: Implications in brain-related diseases. World Journal of Biological Chemistry. 2014, 5, 409–409. [Google Scholar] [CrossRef]
- Chen, Z.-Y.; Jing, D.; Bath, K.G.; Ieraci, A.; Khan, T.; Siao, C.-J.; Herrera, D.G.; Toth, M.; Yang, C.; McEwen, B.S.; et al. Genetic Variant BDNF (Val66Met) Polymorphism Alters Anxiety-Related Behavior. Science. 2006, 314, 140–143. [Google Scholar] [CrossRef]
- Miller, S.; Hallmayer, J.; Wang, P.W.; Hill, S.J.; Johnson, S.L.; Ketter, T.A. Brain-derived neurotrophic factor val66met genotype and early life stress effects upon bipolar course. J. Psychiatr. Res. 2013, 47, 252–258. [Google Scholar] [CrossRef]
- Daskalakis, N.P.; De Kloet, E.R.; Yehuda, R.; Malaspina, D.; Kranz, T.M. Early Life Stress Effects on Glucocorticoid-BDNF Interplay in the Hippocampus. Front. Mol. Neurosci. 2015, 8, 68. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.; Chen, L.; Yang, J.; Han, D.; Fang, D.; Qiu, X.; Yang, X.; Qiao, Z.; Ma, J.; Wang, L.; et al. BDNF Val66Met polymorphism, life stress and depression: A meta-analysis of gene-environment interaction. Journal of Affective Disorders. 2018, 227, 226–235. [Google Scholar] [CrossRef] [PubMed]
- Caldieraro, M.A.; McKee, M.; Leistner-Segal, S.; Vares, E.A.; Kubaski, F.; Spanemberg, L.; Brusius-Facchin, A.C.; Fleck, M.P.; Mischoulon, D. Val66Met polymorphism association with serum BDNF and inflammatory biomarkers in major depression. The World Journal of Biological Psychiatry. 2018, 19, 402–409. [Google Scholar] [CrossRef] [PubMed]
- Lotrich, F.E.; Albusaysi, S.; Ferrell, R.E. Brain-Derived Neurotrophic Factor Serum Levels and Genotype: Association with Depression during Interferon-α Treatment. Neuropsychopharmacology. 2013, 38, 985–995. [Google Scholar] [CrossRef]
- Dooley, L.N.; Ganz, P.A.; Cole, S.W.; Crespi, C.M.; Bower, J.E. Val66Met BDNF polymorphism as a vulnerability factor for inflammation-associated depressive symptoms in women with breast cancer. Journal of Affective Disorders. 2016, 197, 43–50. [Google Scholar] [CrossRef]
- Lindholm, J.S.; Castren, E. Mice with altered BDNF signaling as models for mood disorders and antidepressant effects. Front. Behav. Neurosci. 2014, 8, 143. [Google Scholar] [CrossRef] [PubMed]
- Parrott, J.M.; Redus, L.; Santana-Coelho, D.; Morales, J.; Gao, X.; O'Connor, J.C. Neurotoxic kynurenine metabolism is increased in the dorsal hippocampus and drives distinct depressive behaviors during inflammation. Translational Psychiatry. 2016, 6, e918–e918. [Google Scholar] [CrossRef] [PubMed]
- Laumet, G.; Zhou, W.; Dantzer, R.; Edralin, J.D.; Huo, X.; Budac, D.P.; O'Connor, J.C.; Lee, A.W.; Heijnen, C.J.; Kavelaars, A. Upregulation of neuronal kynurenine 3-monooxygenase mediates depression-like behavior in a mouse model of neuropathic pain. Brain, Behavior, and Immunity. 2017, 66, 94–102. [Google Scholar] [CrossRef]
- Johnson, D.R.; O'Connor, J.C.; Hartman, M.E.; Tapping, R.I.; Freund, G.G. Acute Hypoxia Activates the Neuroimmune System, Which Diabetes Exacerbates. The Journal of Neuroscience. 2007, 27, 1161–1166. [Google Scholar] [CrossRef]
- Sarnyai, Z.; Sibille, E.L.; Pavlides, C.; Fenster, R.J.; McEwen, B.S.; Tóth, M. Impaired hippocampal-dependent learning and functional abnormalities in the hippocampus in mice lacking serotonin <sub>1A</sub> receptors. Proceedings of the National Academy of Sciences. 2000, 97, 14731–14736. [Google Scholar]
- Bussey, T.J.; Padain, T.L.; Skillings, E.A.; Winters, B.D.; Morton, A.J.; Saksida, L.M. The touchscreen cognitive testing method for rodents: How to get the best out of your rat. Learning & Memory. 2008, 15, 516–523. [Google Scholar]
- Paxinos, G.; Franklin, K.B.J. Paxinos and Franklin's the Mouse Brain in Stereotaxic Coordinates. 2012.
- Schmittgen, T.D.; Livak, K.J. Analyzing real-time PCR data by the comparative CT method. Nature Protocols. 2008, 3, 1101–1108. [Google Scholar] [CrossRef]
- Heisler, J.M.; O’Connor, J.C. Indoleamine 2,3-dioxygenase-dependent neurotoxic kynurenine metabolism mediates inflammation-induced deficit in recognition memory. Brain, Behavior, and Immunity. 2015, 50, 115–124. [Google Scholar] [CrossRef]
- Sapolsky, R.M. Depression, antidepressants, and the shrinking hippocampus. Proceedings of the National Academy of Sciences. 2001, 98, 12320–12322. [Google Scholar] [CrossRef]
- Heshmati, M.; Russo, S.J. Anhedonia and the Brain Reward Circuitry in Depression. Current Behavioral Neuroscience Reports. 2015, 2, 146–153. [Google Scholar] [CrossRef]
- Shirayama, Y.; Chaki, S. Neurochemistry of the Nucleus Accumbens and its Relevance to Depression and Antidepressant Action in Rodents. Current Neuropharmacology. 2006, 4, 277–291. [Google Scholar] [CrossRef]
- Gourley, S.L.; Kiraly, D.D.; Howell, J.L.; Olausson, P.; Taylor, J.R. Acute Hippocampal Brain-Derived Neurotrophic Factor Restores Motivational and Forced Swim Performance After Corticosterone. Biological Psychiatry. 2008, 64, 884–890. [Google Scholar] [CrossRef] [PubMed]
- Gyekis, J.P.; Yu, W.; Dong, S.; Wang, H.; Qian, J.; Kota, P.; Yang, J. No association of genetic variants in BDNF with major depression: A meta- and gene-based analysis. Am J Med Genet B Neuropsychiatr Genet. 2013, 162B, 61–70. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Chang, H.; Xiao, X. BDNF Val66Met polymorphism and bipolar disorder in European populations: A risk association in case-control, family-based and GWAS studies. Neurosci. Biobehav. Rev. 2016, 68, 218–233. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Wang, D.D.; Wang, Y.; Liu, T.; Lee, F.S.; Chen, Z.Y. Variant brain-derived neurotrophic factor Val66Met polymorphism alters vulnerability to stress and response to antidepressants. J. Neurosci. 2012, 32, 4092–4101. [Google Scholar] [CrossRef]
- Chen, J.; Li, X.; McGue, M. Interacting effect of BDNF Val66Met polymorphism and stressful life events on adolescent depression. Genes. Brain Behav. 2012, 11, 958–965. [Google Scholar] [CrossRef]
- Notaras, M.; Du, X.; Gogos, J.; van den Buuse, M.; Hill, R.A. The BDNF Val66Met polymorphism regulates glucocorticoid-induced corticohippocampal remodeling and behavioral despair. Transl. Psychiatry. 2017, 7, e1233. [Google Scholar] [CrossRef]
- O'Connor, J.C.; Lawson, M.A.; Andre, C.; Moreau, M.; Lestage, J.; Castanon, N.; Kelley, K.W.; Dantzer, R. Lipopolysaccharide-induced depressive-like behavior is mediated by indoleamine 2,3-dioxygenase activation in mice. Mol. Psychiatry. 2009, 14, 511–522. [Google Scholar] [CrossRef]
- Capuron, L.; Miller, A.H. Immune system to brain signaling: Neuropsychopharmacological implications. Pharmacology & Therapeutics. 2011, 130, 226–238. [Google Scholar]
- Felger, J.C.; Miller, A.H. Cytokine effects on the basal ganglia and dopamine function: The subcortical source of inflammatory malaise. Front. Neuroendocrinol. 2012, 33, 315–327. [Google Scholar] [CrossRef]
- Felger, J.C.; Lotrich, F.E. Inflammatory cytokines in depression: Neurobiological mechanisms and therapeutic implications. Neuroscience. 2013, 246, 199–229. [Google Scholar] [CrossRef]
- Bekhbat, M.; Treadway, M.T.; Felger, J.C. Inflammation as a Pathophysiologic Pathway to Anhedonia: Mechanisms and Therapeutic Implications. Curr. Top. Behav. Neurosci. 2022, 58, 397–419. [Google Scholar] [PubMed]
- Dantzer, R.; O'Connor, J.C.; Freund, G.G.; Johnson, R.W.; Kelley, K.W. From inflammation to sickness and depression: When the immune system subjugates the brain. Nat. Rev. Neurosci. 2008, 9, 46–56. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Yao, T.; Cai, J.; Fu, X.; Li, H.; Wu, J. Systemic inflammatory regulators and 7 major psychiatric disorders: A two-sample Mendelian randomization study. Prog. Neuropsychopharmacol. Biol. Psychiatry. 2022, 116, 110534. [Google Scholar] [CrossRef] [PubMed]
- Banks, W.A.; Kastin, A.J.; Gutierrez, E.G. Penetration of interleukin-6 across the murine blood-brain barrier. Neuroscience Letters. 1994, 179, 53–56. [Google Scholar] [CrossRef]
- Goehler, L.E.; Relton, J.K.; Dripps, D.; Kiechle, R.; Tartaglia, N.; Maier, S.F.; Watkins, L.R. Vagal Paraganglia Bind Biotinylated Interleukin-1 Receptor Antagonist: A Possible Mechanism for Immune-to-Brain Communication. Brain Research Bulletin. 1997, 43, 357–364. [Google Scholar] [CrossRef]
- Wohleb, E.S.; McKim, D.B.; Sheridan, J.F.; Godbout, J.P. Monocyte trafficking to the brain with stress and inflammation: A novel axis of immune-to-brain communication that influences mood and behavior. Front. Neurosci. 2014, 8, 447. [Google Scholar] [CrossRef]
- Reader, B.F.; Jarrett, B.L.; McKim, D.B.; Wohleb, E.S.; Godbout, J.P.; Sheridan, J.F. Peripheral and central effects of repeated social defeat stress: Monocyte trafficking, microglial activation, and anxiety. Neuroscience. 2015, 289, 429–442. [Google Scholar] [CrossRef]
- McKim, D.B.; Yin, W.; Wang, Y.; Cole, S.W.; Godbout, J.P.; Sheridan, J.F. Social Stress Mobilizes Hematopoietic Stem Cells to Establish Persistent Splenic Myelopoiesis. Cell Rep. 2018, 25, 2552–2562. [Google Scholar] [CrossRef]
- McKim, D.B.; Weber, M.D.; Niraula, A.; Sawicki, C.M.; Liu, X.; Jarrett, B.L.; Ramirez-Chan, K.; Wang, Y.; Roeth, R.M.; Sucaldito, A.D.; et al. Microglial recruitment of IL-1β-producing monocytes to brain endothelium causes stress-induced anxiety. Molecular Psychiatry. 2018, 23, 1421–1431. [Google Scholar] [CrossRef]
- Menard, C.; Pfau, M.L.; Hodes, G.E.; Kana, V.; Wang, V.X.; Bouchard, S.; Takahashi, A.; Flanigan, M.E.; Aleyasin, H.; LeClair, K.B.; et al. Social stress induces neurovascular pathology promoting depression. Nat. Neurosci. 2017, 20, 1752–1760. [Google Scholar] [CrossRef]
- Gaire, S.; An, J.; Yang, H.; Lee, K.A.; Dumre, M.; Lee, E.J.; Park, S.M.; Joe, E.H. Systemic inflammation attenuates the repair of damaged brains through reduced phagocytic activity of monocytes infiltrating the brain. Mol. Brain. 2024, 17, 47. [Google Scholar] [CrossRef]
- Cazareth, J.; Guyon, A.; Heurteaux, C.; Chabry, J.; Petit-Paitel, A. Molecular and cellular neuroinflammatory status of mouse brain after systemic lipopolysaccharide challenge: Importance of CCR2/CCL2 signaling. Journal of Neuroinflammation. 2014, 11, 132. [Google Scholar] [CrossRef] [PubMed]
- Bodea, L.G.; Wang, Y.; Linnartz-Gerlach, B.; Kopatz, J.; Sinkkonen, L.; Musgrove, R.; Kaoma, T.; Muller, A.; Vallar, L.; Di Monte, D.A.; et al. Neurodegeneration by activation of the microglial complement-phagosome pathway. J. Neurosci. 2014, 34, 8546–8556. [Google Scholar] [CrossRef] [PubMed]
- Yeo, H.G.; Hong, J.J.; Lee, Y.; Yi, K.S.; Jeon, C.Y.; Park, J.; Won, J.; Seo, J.; Ahn, Y.J.; Kim, K.; et al. Increased CD68/TGFbeta Co-expressing Microglia/ Macrophages after Transient Middle Cerebral Artery Occlusion in Rhesus Monkeys. Exp. Neurobiol. 2019, 28, 458–473. [Google Scholar] [CrossRef] [PubMed]
- Chistiakov, D.A.; Killingsworth, M.C.; Myasoedova, V.A.; Orekhov, A.N.; Bobryshev, Y.V. CD68/macrosialin: Not just a histochemical marker. Lab. Invest. 2017, 97, 4–13. [Google Scholar] [CrossRef]
- Duan, N.; Zhang, Y.; Tan, S.; Sun, J.; Ye, M.; Gao, H.; Pu, K.; Wu, M.; Wang, Q.; Zhai, Q. Therapeutic targeting of STING-TBK1-IRF3 signalling ameliorates chronic stress induced depression-like behaviours by modulating neuroinflammation and microglia phagocytosis. Neurobiol. Dis. 2022, 169, 105739. [Google Scholar] [CrossRef]
- Feng, X.; Fan, Y.; Chung, C.Y. Mefenamic acid can attenuate depressive symptoms by suppressing microglia activation induced upon chronic stress. Brain Res. 2020, 1740, 146846. [Google Scholar] [CrossRef]
- Zhu, Y.; Haddad, Y.; Yun, H.J.; Geng, X.; Ding, Y. Induced Inflammatory and Oxidative Markers in Cerebral Microvasculature by Mentally Depressive Stress. Mediators Inflamm. 2023, 2023, 4206316. [Google Scholar] [CrossRef]
- Sandrini, L.; Castiglioni, L.; Amadio, P.; Werba, J.P.; Eligini, S.; Fiorelli, S.; Zara, M.; Castiglioni, S.; Bellosta, S.; Lee, F.S.; et al. Impact of BDNF Val66Met Polymorphism on Myocardial Infarction: Exploring the Macrophage Phenotype. Cells. 2020, 9. [Google Scholar] [CrossRef]
- Gray, J.D.; Rubin, T.G.; Kogan, J.F.; Marrocco, J.; Weidmann, J.; Lindkvist, S.; Lee, F.S.; Schmidt, E.F.; McEwen, B.S. Translational profiling of stress-induced neuroplasticity in the CA3 pyramidal neurons of BDNF Val66Met mice. Mol. Psychiatry. 2018, 23, 904–913. [Google Scholar] [CrossRef]
- Jing, D.; Lee, F.S.; Ninan, I. The BDNF Val66Met polymorphism enhances glutamatergic transmission but diminishes activity-dependent synaptic plasticity in the dorsolateral striatum. Neuropharmacology. 2017, 112, 84–93. [Google Scholar] [CrossRef] [PubMed]
- Huwart, S.J.P.; Fayt, C.; Gangarossa, G.; Luquet, S.; Cani, P.D.; Everard, A. TLR4-dependent neuroinflammation mediates LPS-driven food-reward alterations during high-fat exposure. J. Neuroinflammation. 2024, 21, 305. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Lai, S.; Zhou, T.; Xia, Z.; Li, W.; Sha, W.; Liu, J.; Chen, Y. Progranulin from different gliocytes in the nucleus accumbens exerts distinct roles in FTD- and neuroinflammation-induced depression-like behaviors. J. Neuroinflammation. 2022, 19, 318. [Google Scholar] [CrossRef]
- Wang, J.; Lai, S.; Wang, R.; Zhou, T.; Dong, N.; Zhu, L.; Chen, T.; Zhang, X.; Chen, Y. Dopamine D3 receptor in the nucleus accumbens alleviates neuroinflammation in a mouse model of depressive-like behavior. Brain Behav. Immun. 2022, 101, 165–179. [Google Scholar] [CrossRef]
- Maier, S.F.; Seligman, M.E. Learned helplessness at fifty: Insights from neuroscience. Psychol. Rev. 2016, 123, 349–367. [Google Scholar] [CrossRef]
- Dincheva, I.; Yang, J.; Li, A.; Marinic, T.; Freilingsdorf, H.; Huang, C.; Casey, B.J.; Hempstead, B.; Glatt, C.E.; Lee, F.S.; et al. Effect of Early-Life Fluoxetine on Anxiety-Like Behaviors in BDNF Val66Met Mice. Am. J. Psychiatry. 2017, 174, 1203–1213. [Google Scholar] [CrossRef]
- Bath, K.G.; Jing, D.Q.; Dincheva, I.; Neeb, C.C.; Pattwell, S.S.; Chao, M.V.; Lee, F.S.; Ninan, I. BDNF Val66Met impairs fluoxetine-induced enhancement of adult hippocampus plasticity. Neuropsychopharmacology. 2012, 37, 1297–1304. [Google Scholar] [CrossRef]
- Hao, R.; Qi, Y.; Hou, D.N.; Ji, Y.Y.; Zheng, C.Y.; Li, C.Y.; Yung, W.H.; Lu, B.; Huang, Y. BDNF val66met Polymorphism Impairs Hippocampal Long-Term Depression by Down-Regulation of 5-HT3 Receptors. Front. Cell Neurosci. 2017, 11, 306. [Google Scholar]
- Mallei, A.; Ieraci, A.; Corna, S.; Tardito, D.; Lee, F.S.; Popoli, M. Global epigenetic analysis of BDNF Val66Met mice hippocampus reveals changes in dendrite and spine remodeling genes. Hippocampus. 2018, 28, 783–795. [Google Scholar] [CrossRef]
- Ninan, I.; Bath, K.G.; Dagar, K.; Perez-Castro, R.; Plummer, M.R.; Lee, F.S.; Chao, M.V. The BDNF Val66Met polymorphism impairs NMDA receptor-dependent synaptic plasticity in the hippocampus. J. Neurosci. 2010, 30, 8866–8870. [Google Scholar] [CrossRef] [PubMed]
- Reichenberg, A.; Yirmiya, R.; Schuld, A.; Kraus, T.; Haack, M.; Morag, A.; Pollmächer, T. Cytokine-Associated Emotional and Cognitive Disturbances in Humans. Archives of General. Psychiatry. 2001, 58, 445–445. [Google Scholar] [CrossRef]
- Cohen, S.; Janicki-Deverts, D.; Doyle, W.J.; Miller, G.E.; Frank, E.; Rabin, B.S.; Turner, R.B. Chronic stress, glucocorticoid receptor resistance, inflammation, and disease risk. Proc. Natl. Acad. Sci. U S A. 2012, 109, 5995–5999. [Google Scholar] [CrossRef] [PubMed]
- Mac Giollabhui, N.; Ng, T.H.; Ellman, L.M.; Alloy, L.B. The longitudinal associations of inflammatory biomarkers and depression revisited: Systematic review, meta-analysis, and meta-regression. Mol Psychiatry. 2021, 26, 3302–3314. [Google Scholar] [CrossRef] [PubMed]
- van Eeden, W.A.; van Hemert, A.M.; Carlier, I.V.E.; Penninx, B.; Lamers, F.; Fried, E.I.; Schoevers, R.; Giltay, EJ. Basal and LPS-stimulated inflammatory markers and the course of individual symptoms of depression. Transl. Psychiatry. 2020, 10, 235. [Google Scholar] [CrossRef]
- Can, A.; Dao, D.T.; Arad, M.; Terrillion, C.E.; Piantadosi, S.C.; Gould, T.D. The mouse forced swim test. J Vis Exp. 2012, e3638. [Google Scholar] [CrossRef]
- Bogdanova, O.V.; Kanekar, S.; D'Anci, K.E.; Renshaw, P.F. Factors influencing behavior in the forced swim test. Physiol. Behav. 2013, 118, 227–239. [Google Scholar] [CrossRef]
- Anyan, J.; Amir, S. Too Depressed to Swim or Too Afraid to Stop? A Reinterpretation of the Forced Swim Test as a Measure of Anxiety-Like Behavior. Neuropsychopharmacology. 2018, 43, 931–933. [Google Scholar] [CrossRef]
- Montag, C.; Basten, U.; Stelzel, C.; Fiebach, C.J.; Reuter, M. The BDNF Val66Met polymorphism and anxiety: Support for animal knock-in studies from a genetic association study in humans. Psychiatry Research. 2010, 179, 86–90. [Google Scholar] [CrossRef]
- Negron, M.; Kristensen, J.; Nguyen, V.T.; Gansereit, L.E.; Raucci, F.J.; Chariker, J.L.; Heck, A.; Brula, I.; Kitchen, G.; Awgulewitsch, C.P.; et al. Sex-Based Differences in Cardiac Gene Expression and Function in BDNF Val66Met Mice. Int J Mol Sci. 2021, 22. [Google Scholar]
- Bath, K.G.; Chuang, J.; Spencer-Segal, J.L.; Amso, D.; Altemus, M.; McEwen, B.S.; Lee, F.S. Variant brain-derived neurotrophic factor (Valine66Methionine) polymorphism contributes to developmental and estrous stage-specific expression of anxiety-like behavior in female mice. Biol. Psychiatry. 2012, 72, 499–504. [Google Scholar] [CrossRef]
- Miranda, M.; Morici, J.F.; Zanoni, M.B.; Bekinschtein, P. Brain-Derived Neurotrophic Factor: A Key Molecule for Memory in the Healthy and the Pathological Brain. Front. Cell Neurosci. 2019, 13, 363. [Google Scholar] [CrossRef]
Figure 1.
Experimental Design and Timeline - Three days prior to LPS injection, mice were trained for sucrose preference. Two hours after LPS, locomotion and social exploratory behavior were measured, and plasma and brains were collected. Twelve hours after LPS, the whole brain was collected. 24 hours following LPS, locomotor activity, social exploration, depressive-like behavior, and working memory were measured. Then plasma, brain, hippocampus, nucleus accumbens were collected.
Figure 1.
Experimental Design and Timeline - Three days prior to LPS injection, mice were trained for sucrose preference. Two hours after LPS, locomotion and social exploratory behavior were measured, and plasma and brains were collected. Twelve hours after LPS, the whole brain was collected. 24 hours following LPS, locomotor activity, social exploration, depressive-like behavior, and working memory were measured. Then plasma, brain, hippocampus, nucleus accumbens were collected.
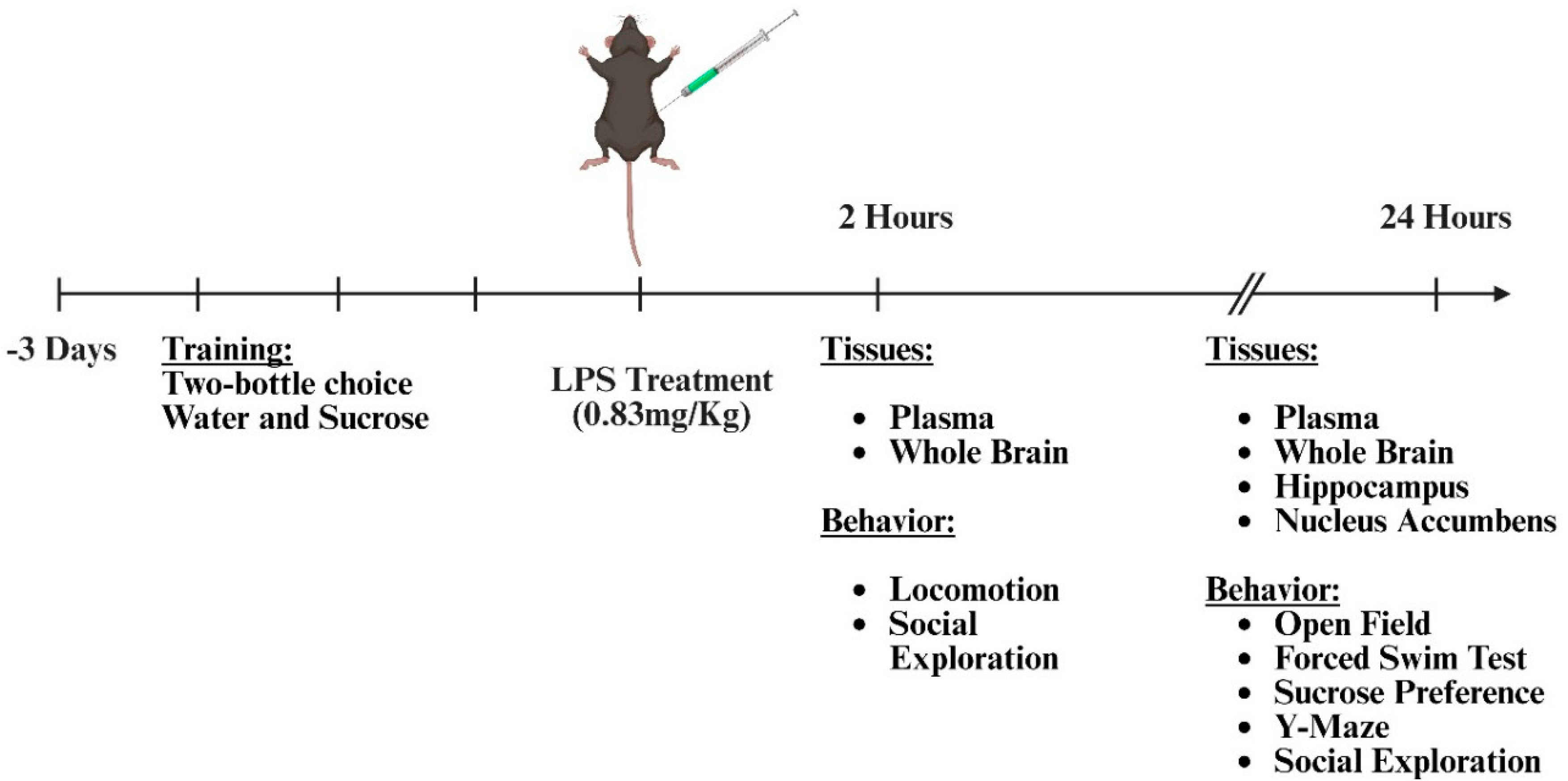
Figure 2.
BDNF Val66Met expression results in susceptibility to the anhedonia-like behavioral effects of immune challenge. 24 hours after intraperitoneal saline or LPS administration, the following metrics were measured, and data were analyzed using a two-way ANOVA with genotype and LPS as independent factors for normally distributed data and non-parametric Kruskal-Wallis’s test were used to analyze non-normally distributed data. A) Sucrose preference; dotted line is at the 50% mark, which indicates no preference (n=10 mice/group); Two-way ANOVA interaction – genotype x LPS: F (1,36) = 6.285, p=0.0168, η2p = 0.149. B) Forced swim test (Kruskal-Wallis’s test, p=0.6397, n=6-7 mice/group). C) Open field test (n=5-7 mice/group); main effect of LPS: F (1,20) = 6.088, p=0.0228, η2p = 0.407. D) Discrimination ratio in Y-maze working memory task (n=8-10 mice/group). E) Change in body weight (n=13-17 mice/group); main effect of LPS: F (1,56) = 146.2, p<0.0001, η2p = 0.723. F) Social exploratory behavior (n=9-11 mice/group). Error bars represent +/- standard error of means (SEM). Post-hoc testing with Tukey’s multiple comparison test to compare differences between group means if main effects of genotype or LPS were detected and for non-normally distributed data post-hoc testing with corrected Dunn’s multiple comparison test was used to compare differences between group means; # represents main effect of LPS; ns - not significant; *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001.
Figure 2.
BDNF Val66Met expression results in susceptibility to the anhedonia-like behavioral effects of immune challenge. 24 hours after intraperitoneal saline or LPS administration, the following metrics were measured, and data were analyzed using a two-way ANOVA with genotype and LPS as independent factors for normally distributed data and non-parametric Kruskal-Wallis’s test were used to analyze non-normally distributed data. A) Sucrose preference; dotted line is at the 50% mark, which indicates no preference (n=10 mice/group); Two-way ANOVA interaction – genotype x LPS: F (1,36) = 6.285, p=0.0168, η2p = 0.149. B) Forced swim test (Kruskal-Wallis’s test, p=0.6397, n=6-7 mice/group). C) Open field test (n=5-7 mice/group); main effect of LPS: F (1,20) = 6.088, p=0.0228, η2p = 0.407. D) Discrimination ratio in Y-maze working memory task (n=8-10 mice/group). E) Change in body weight (n=13-17 mice/group); main effect of LPS: F (1,56) = 146.2, p<0.0001, η2p = 0.723. F) Social exploratory behavior (n=9-11 mice/group). Error bars represent +/- standard error of means (SEM). Post-hoc testing with Tukey’s multiple comparison test to compare differences between group means if main effects of genotype or LPS were detected and for non-normally distributed data post-hoc testing with corrected Dunn’s multiple comparison test was used to compare differences between group means; # represents main effect of LPS; ns - not significant; *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001.
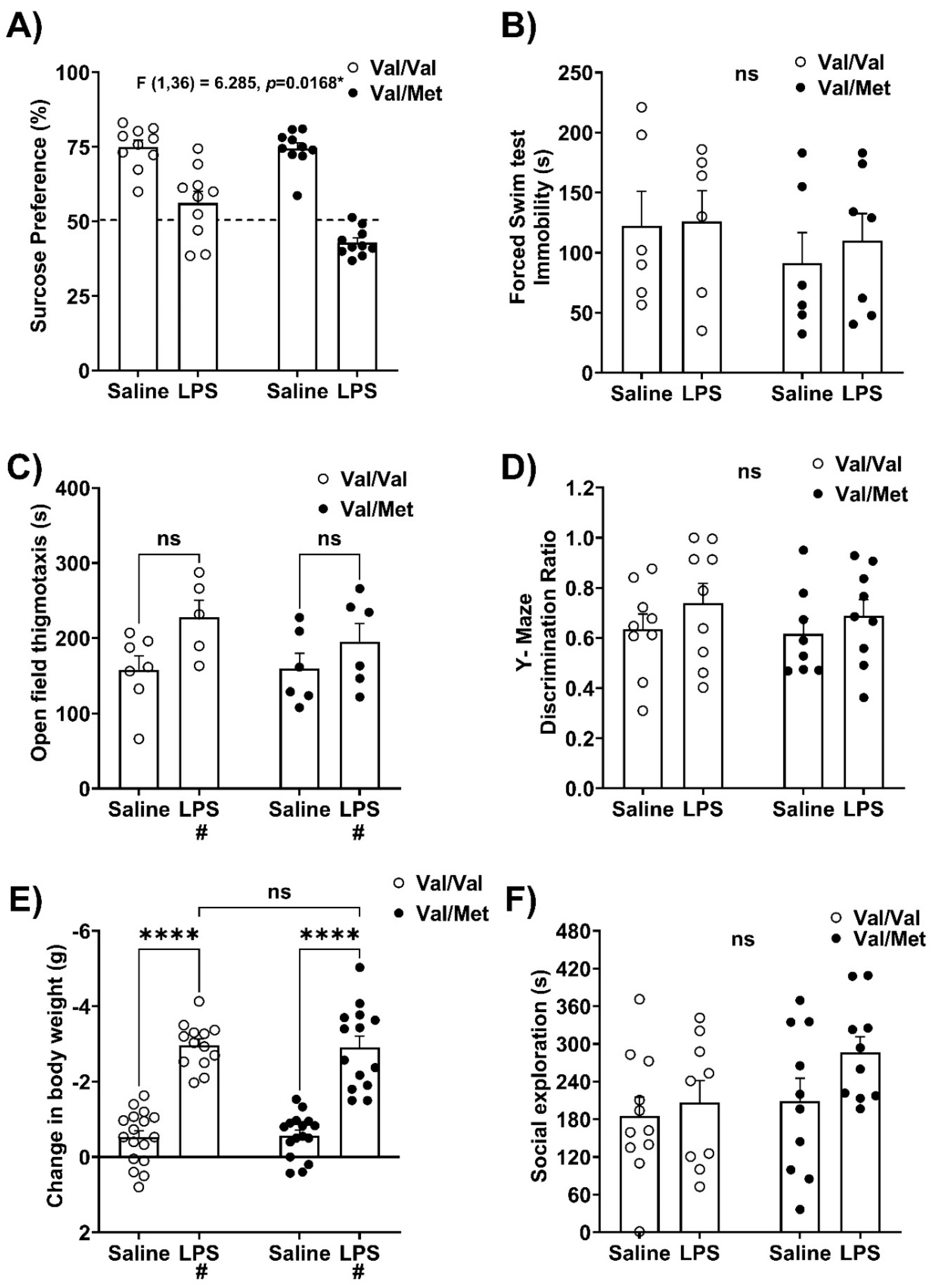
Figure 3.
Immune challenge induces dysregulated whole brain pro-inflammatory cytokine expression in BDNF Val66Met Mice. After 2 or 24 hours of saline or LPS administration, the following PCR targets were measured from whole brain, and data were analyzed using a two-way ANOVA with genotype and LPS as independent factors for normally distributed data and non-parametric Kruskal-Wallis’s test were used to analyze non-normally distributed data. The mRNA expression of A) mRNA expression of Interleukin-1β (IL-1β), main effects of LPS: F (1,10) = 36.28, p=0.0001, η2p = 0.784; B) Tumor necrosis factor-alpha (TNFα); Kruskal-Wallis’s test H (3) = 12.63, p=0.0055, η2 = 0.574 at 2 hours following intraperitoneal saline or LPS administration. The mRNA expression of D) IL-1β, Kruskal-Wallis’s test H (3) = 19.85, p=0.0002, η2 = 0.735; E) TNFα, genotype x LPS interaction: F (1,21) = 4.512, p=0.0457, η2p = 0.177, at 24 hours following intraperitoneal saline or LPS administration. n=3-9 mice/group/timepoint. Error bars represent +/- standard error of means (SEM). Post-hoc testing with Tukey’s multiple comparison test to compare differences between group means if main effects of genotype or LPS were detected and for non-normally distributed data post-hoc testing with corrected Dunn’s multiple comparison test was used to compare differences between group means; # represents main effect of LPS; ns - not significant; *p<0.05, **p<0.01, ***p<0.001.
Figure 3.
Immune challenge induces dysregulated whole brain pro-inflammatory cytokine expression in BDNF Val66Met Mice. After 2 or 24 hours of saline or LPS administration, the following PCR targets were measured from whole brain, and data were analyzed using a two-way ANOVA with genotype and LPS as independent factors for normally distributed data and non-parametric Kruskal-Wallis’s test were used to analyze non-normally distributed data. The mRNA expression of A) mRNA expression of Interleukin-1β (IL-1β), main effects of LPS: F (1,10) = 36.28, p=0.0001, η2p = 0.784; B) Tumor necrosis factor-alpha (TNFα); Kruskal-Wallis’s test H (3) = 12.63, p=0.0055, η2 = 0.574 at 2 hours following intraperitoneal saline or LPS administration. The mRNA expression of D) IL-1β, Kruskal-Wallis’s test H (3) = 19.85, p=0.0002, η2 = 0.735; E) TNFα, genotype x LPS interaction: F (1,21) = 4.512, p=0.0457, η2p = 0.177, at 24 hours following intraperitoneal saline or LPS administration. n=3-9 mice/group/timepoint. Error bars represent +/- standard error of means (SEM). Post-hoc testing with Tukey’s multiple comparison test to compare differences between group means if main effects of genotype or LPS were detected and for non-normally distributed data post-hoc testing with corrected Dunn’s multiple comparison test was used to compare differences between group means; # represents main effect of LPS; ns - not significant; *p<0.05, **p<0.01, ***p<0.001.
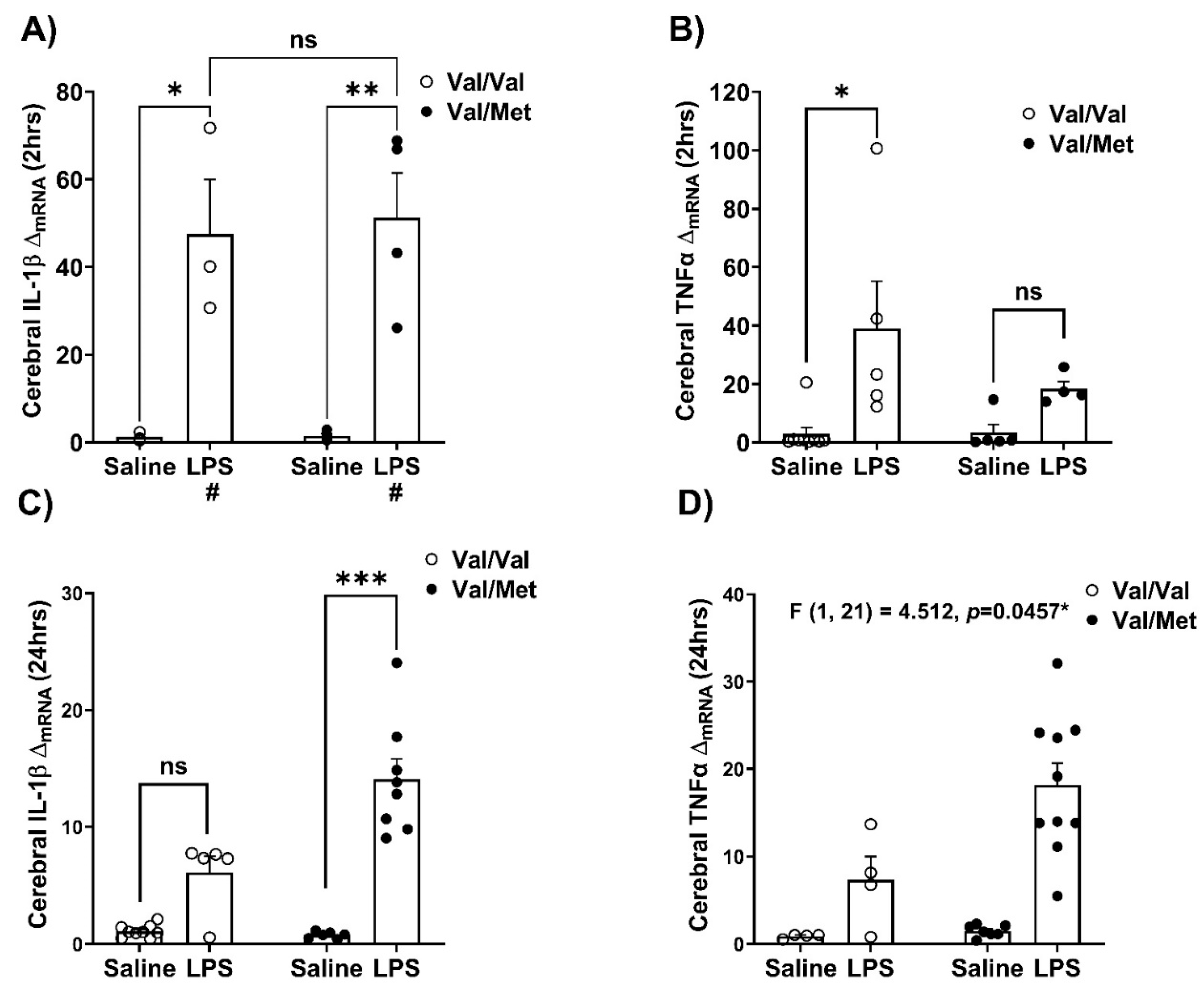
Figure 4.
Measurement of pro-inflammatory factors in the hippocampus in Val66Val and Val66Met mice in response to peripheral immune activation. After 24 hours, whole brains were micro-dissected and PCR was performed to determine relative gene expression in the hippocampus, and data were analyzed with the non-parametric Kruskal-Wallis’s test. The mRNA expression of A) Kruskal-Wallis’s test H (3) = 12.63, p=0.0055, η2 = 0.486; B) TNFα, Kruskal-Wallis’s test H (3) = 20.55, p=0.0001, η2 = 0.735, C) CD68, Kruskal-Walli’s test H (3) = 16.22, p=0.0010, η2 = 0.559. n=4-8 mice/group; Post-hoc testing with corrected Dunn’s multiple comparison test was used to compare differences between group means; *p<0.05, **p<0.01, ***p<0.001.
Figure 4.
Measurement of pro-inflammatory factors in the hippocampus in Val66Val and Val66Met mice in response to peripheral immune activation. After 24 hours, whole brains were micro-dissected and PCR was performed to determine relative gene expression in the hippocampus, and data were analyzed with the non-parametric Kruskal-Wallis’s test. The mRNA expression of A) Kruskal-Wallis’s test H (3) = 12.63, p=0.0055, η2 = 0.486; B) TNFα, Kruskal-Wallis’s test H (3) = 20.55, p=0.0001, η2 = 0.735, C) CD68, Kruskal-Walli’s test H (3) = 16.22, p=0.0010, η2 = 0.559. n=4-8 mice/group; Post-hoc testing with corrected Dunn’s multiple comparison test was used to compare differences between group means; *p<0.05, **p<0.01, ***p<0.001.
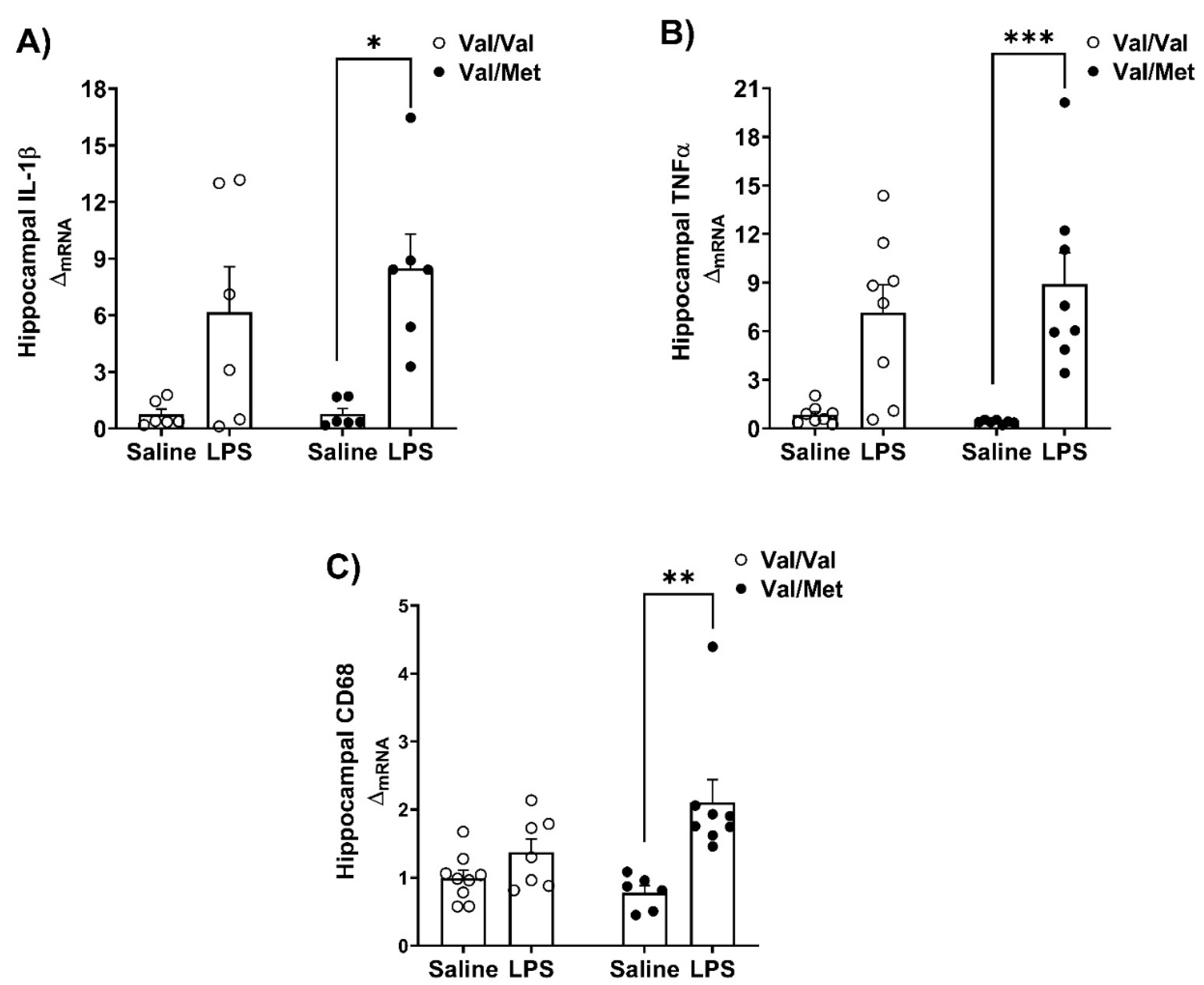

Table 1.
Peripheral immune response is not influenced by BDNF Val66Met polymorphism genotypes. Plasma concentrations of the pro-inflammatory cytokines IL-1β, IL-6, and TNFα at 2 hours (n=3-5 mice/group) and 24 hours (n=6-15 mice/group) after saline or LPS administration, and data were analyzed using a two-way ANOVA with genotype and LPS as independent factors. na – not applicable * p<0.05, **p<0.01, ***p<0.001, ****p<0.0001.
Table 1.
Peripheral immune response is not influenced by BDNF Val66Met polymorphism genotypes. Plasma concentrations of the pro-inflammatory cytokines IL-1β, IL-6, and TNFα at 2 hours (n=3-5 mice/group) and 24 hours (n=6-15 mice/group) after saline or LPS administration, and data were analyzed using a two-way ANOVA with genotype and LPS as independent factors. na – not applicable * p<0.05, **p<0.01, ***p<0.001, ****p<0.0001.
| Val/Val | Val/Met | Main Effects | Effect size | ||||||
| Saline | LPS | Saline | LPS | ||||||
| Mean (SEM) | Mean (SEM) | Mean (SEM) | Mean (SEM) | Genotype p-value |
Treatment p-value |
η2p | |||
| 2hrs | |||||||||
| IL-1β | 0.152 (0.152) | 7.596 (6.078) | 0.543 (0.543) | 4.381 (4.381) | 0.2413 | 0.7630 | na | ||
| IL-6 | 1.379 (0.939) | 195.172 (0.0) | 1.500 (0.815) | 152.397 (42.776) | 0.3461 | <0.0001**** | 0.839 | ||
| TNFα | 0.783 (0.783) | 73.288 (19.506) | 11.628 (11.533) | 40.409 (23.830) | 0.5309 | 0.0111* | 0.402 | ||
| 24hrs | |||||||||
| IL-1β | 2.841 (0.998) | 2.315 (1.037) | 3.460 (1.042) | 4.678 (1.903) | 0.8070 | 0.2981 | na | ||
| IL-6 | 0.539 (0.173) | 72.034 (27.041) | 0.956 (0.620) | 75.048 (17.211) | 0.9191 | 0.0001*** | 0.350 | ||
| TNFα | 1.296 (0.746) | 3.292 (1.370) | 0.543 (0.543) | 4.211 (0.644) | 0.9292 | 0.0064** | 0.345 | ||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



