Submitted:
23 June 2025
Posted:
24 June 2025
You are already at the latest version
Abstract
Several inherited connective tissue disorders have been only lately considered in stroke pathogenesis, presenting as part of the phenotype in young adults. Therefore, we present the case of a 24-year-old woman, who suffered from a right occipital ischemic stroke, manifesting with sudden onset of visual disturbance and left body paresthesia. Physical examination revealed blue sclerae, poor wound healing, elastic neck skin, long and thin fingers. Bone density test showed osteoporosis/severe osteopenia. The genetic assessment for juvenile stroke detected C.128delA variant in heterozygosis and apparent mosaicism on the COL1A gene, involved in Overlap Ehlers-Danlos syndrome (EDS)/ Osteogenesis imperfecta (OI), and c.1195A> G variant in heterozygosis on TGFB2 gene, involved in Loeys-Dietz syndrome (LDS). Familial aggregation study revealed the very same variant on TGFB2 gene in the patient’s daughter. Considering the coexistence of both diseases, we believe that stroke may correlate to LDS, while phenotypic picture is related to the overlap EDS/OI mosaicism.
Keywords:
connective tissue disorders
; stroke in young adults
; Loeys-Dietz syndrome (LDS)
; Overlap Ehlers-Danlos syndrome (EDS)/ Osteogenesis imperfecta (OI)
; C.128delA variant on the COL1A gene
; c.1195A>G variant on TGFB2 gene
1. Introduction
The incidence of stroke increases exponentially with age, but it may also occur in children and young adults, resulting in significant morbidity and mortality. The identification of stroke cause in young adults is mostly challenging unlike in the elderly, as cerebrovascular accident in young adults can be more often due to rare causes and infrequent risk factors [1]. Next to arteriopathies or metabolic diseases, several inherited connective tissue disorders (CTD) may present ischemic or hemorrhagic stroke as part of the phenotype in young adults. While some of CTD have been recognized for many years as causes of stroke, others have been lately considered in stroke pathogenesis [2]. Specifically, the incidence of Ehlers-Danlos syndrome (EDS) is best estimated to be between 1 in 2500 and 1 in 5000, including all subtypes in the general population [3], whereas the vascular type is the rarest form, affecting about 1 in 250,000 people worldwide [4]. Furthermore, the combination of EDS and osteogenesis imperfecta (OI) is very rare (< 1/1,000,000) [5,6]. On the other hand, Loeys-Dietz syndrome (LDS) is considered a rare genetic connective tissue disorder, but prevalence and incidence of the disease are currently unknown [7,8]. Guidelines and recommendations are lacking in the diagnostic process and clinical approach for these patients. We present the following case report in order to underline the rarity of young adult ischemic stroke causes and the diagnostic difficulties.
2. Clinical Presentation
A 24-year-old woman, with a personal history of knee dislocation and mild scoliosis, suffered from a right occipital ischemic stroke (Figure 1), manifesting with sudden onset of visual disturbance and left body paresthesia. At that time, the patient started the single antiplatelet therapy (aspirin 100mg) and was discharged as minor stroke of undetermined cause [9], persisting a residual left-sided incomplete homonymous hemianopia (National Institutes of Health Stroke Scale – NIHSS: 1). After six-year follow-up with serial radiological evaluation excluding recurrences, she was finally admitted to stroke department for in-depth assessment. Herein, laboratory and instrumental tests for stroke cause identification were performed in outpatient setting: supra-aortic vessels and transcranial Doppler ultrasound, 24-hour electrocardiogram monitoring, transthoracic and transesophageal echocardiography, brain computed tomography (CT) and magnetic resonance angiography (MRA) (Figure 2), and autoantibody panel for autoimmune disease. In addition, a thorough genetic screening included hereditary thrombophilia testing performed by dot blot technique, lysosomal storage and mitochondrial diseases, and a molecular analysis of a panel of genes associated to juvenile stroke assessed with next-generation sequencing (NGS).
3. Results
The instrumental assessment did not reveal large vessels atherosclerosis, cardiovascular diseases or brain vessel abnormalities. Antinuclear antibodies (ANA) with coarse speckled pattern, lupus anticoagulant (LAC) and immunoglobulin G subclass of antiphospholipid antibodies resulted positive after a single measurement. Repeated assessment did not confirm these findings, but Basedow thyroiditis was later diagnosed. Genetic panel for Fabry disease, mitochondrial myopathy, encephalopathy, lactic acidosis, and stroke-like episodes (MELAS), and myoclonic epilepsy with ragged-red fibers (MERRF) was unremarkable. Nevertheless, the study of genes associated with juvenile stroke detected C.128delA variant in heterozygosity and apparent mosaicism on the COL1A gene and c.1195A> G variant in heterozygosity on TGFB2 gene. Physical examination revealed blue sclerae, poor wound healing, elastic neck skin, long and thin fingers. Bone mineral density test showed osteoporosis at the rachis and severe osteopenia at the femoral neck.
Familial Aggregation Study performed on the patient’s daughter revealed the presence of the very same variant on TGFB2 gene.
4. Discussion
Considering the clinical features and the genetic findings, we diagnosed the coexistence of LDS and EDS/OI overlap syndrome in mosaicism.
EDS/OI overlap syndrome is a recently described disorder of connective tissue, characterized by mutation of COL1A1 (17q21.33) or COL1A2 (7q21.3) genes, that are involved in α-1 and α-2 chains of type 1 collagen synthesis 5,6. In this case, we found a C.128delA variant on the COL1A gene, which has never been described in literature but classified as likely pathogenic in VarSome database. The clinical spectrum is broad, as patients may present a mixed phenotype including features of both osteogenesis imperfecta (bone fragility, long bone fractures, blue sclerae, short stature) and Ehlers-Danlos syndrome (joint hyperextensibility, soft and hyperextensible skin, abnormal wound healing, easy bruising, vascular fragility). In the case described, we found also a c.1195A>G variant on TGFB2 gene, never described in literature and interpreted as likely pathogenic in VarSome. TGFB2 variants are associated with LDS, an autosomal dominant connective tissue disorder primarily characterized by vascular and skeletal involvement, including arterial aneurysms and/or dissections, pectus anomalies, scoliosis, joint laxity, and arachnodactyly [7]. Any aneurysms and dissections involving cerebrovascular afferents, as well as carotid and vertebrobasilar systems, may require antiplatelet treatment, aiming to prevent complications such as strokes [1. A skeletal phenotype characterized by low bone mineral density and skeletal fragility (fractures) has been described in some young patients with LDS type 2 (caused by mutation of TGFBR1) [11]. A study by Sponseller and colleagues [12] conducted on individuals with LDS 1/2 revealed a fracture risk of 50% by the age of 14 years. Dual-energy X-ray absorptiometry data revealed in these patients low or very low bone mineral density in the spine, hip and/or femoral neck.
Both variants detected have not been previously described in the literature, but are considered likely pathogenetic. Since these conditions have extremely variable expressiveness, the clinical symptoms of both diseases could occur simultaneously in an unpredictable manner. According to literature, we believe that the IS may be related to LDS, while phenotypic picture is related to the overlap EDS/OI mosaicism.
Considering that this is a case report, the main limitations include the lack of ability to generalize our findings and to establish cause-effect relationship.
5. Conclusions
LDS is characterized by vascular alterations and skeletal manifestations, and the clinical spectrum may be extremely variable, requiring neurological and cardiovascular follow-up. Therefore, stroke of undetermined cause occurring in young adults might require a complete genetic testing for collagen disorders, including Marfan syndrome, OI, EDS, and LDS, in addition to other monogenic diseases.
Author Contributions
: FGi: Drafting/revision of the manuscript for content, including medical writing for content; Major role in the acquisition of data; Study concept or design; Analysis or interpretation of data. FGr: Drafting/revision of the manuscript for content, including medical writing for content; Major role in the acquisition of data. VT: Drafting/revision of the manuscript for content, including medical writing for content. SB: Analysis or interpretation of data. LA: Drafting/revision of the manuscript for content, including medical writing for content. PLS: Study concept or design. AT: Drafting/revision of the manuscript for content, including medical writing for content. RFM: Drafting/revision of the manuscript for content, including medical writing for content; Major role in the acquisition of data; Study concept or design.
Funding
This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
Institutional Review Board Statement
The paper does not report on primary research. All data analyzed were collected as part of routine diagnosis and treatment.
Informed Consent Statement
Informed consent was obtained from all subjects involved in the study. Written informed consent has been obtained from the patient to publish this paper.
Data Availability Statement
Additional unpublished data from the study has not been deposited into a publicly available repository. Data will be made available on request.
Acknowledgments
None declared.
Conflicts of Interest
The authors declare no conflicts of interest.
Abbreviations
The following abbreviations are used in this manuscript:
| CTD | connective tissue disorders |
| EDS | Ehlers-Danlos syndrome |
| OI | osteogenesis imperfecta |
| LDS | Loeys-Dietz syndrome |
| CT | computed tomography |
| MRA | magnetic resonance angiography |
| NIHSS | National Institutes of Health Stroke Scale |
| NGS | next-generation sequencing |
| ANA | Antinuclear antibodies |
| LAC | lupus anticoagulant |
| MELAS | mitochondrial myopathy, encephalopathy, lactic acidosis, and stroke-like episodes |
| MERRF | myoclonic epilepsy with ragged-red fibers |
References
- Putaala, J. Ischemic Stroke in Young Adults. Continuum (Minneap. Minn). 2020, 26, 386–414. [Google Scholar] [CrossRef]
- Vanakker, O. M., Hemelsoet, D. & De Paepe, A. Hereditary connective tissue diseases in young adult stroke: a comprehensive synthesis. Stroke Res. Treat. 2011, (2011).
- Miklovic, T. & Sieg, V. C. Ehlers-Danlos Syndrome. Neurocutaneous Disord. A Clin. Diagnostic Ther. Approach Third Ed. 393–399 (2023). [CrossRef]
- Byers, P. H. et al. Diagnosis, natural history, and management in vascular Ehlers-Danlos syndrome. Am. J. Med. Genet. C. Semin. Med. Genet. 2017, 175, 40–47. [Google Scholar] [CrossRef] [PubMed]
- Morlino, S. et al. COL1-related overlap disorder: A novel connective tissue disorder incorporating the osteogenesis imperfecta/Ehlers-Danlos syndrome overlap: Clinical Genetics. Clin. Genet. 2020, 97, 396–406. [Google Scholar] [CrossRef] [PubMed]
- Morabito, L. A. et al. Osteogenesis Imperfecta/Ehlers-Danlos Overlap Syndrome and Neuroblastoma-Case Report and Review of Literature. Genes (Basel). 13, (2022).
- Loeys, B. L. & Dietz, H. C. Loeys-Dietz Syndrome. GeneReviews® (2018.
- Gouda, P. et al. Clinical features and complications of Loeys-Dietz syndrome: A systematic review. Int. J. Cardiol. 362, 158–167 (2022).
- Adams, H. P. et al. Classification of subtype of acute ischemic stroke. Definitions for use in a multicenter clinical trial. TOAST. Trial of Org 10172 in Acute Stroke Treatment. Stroke 24, 35–41 (1993).
- MacCarrick, G. et al. Loeys–Dietz syndrome: a primer for diagnosis and management. Genet. Med. 16, 576 (2014).
- Kirmani, S. et al. Germline TGF-β receptor mutations and skeletal fragility: A report on two patients with Loeys-Dietz syndrome. Am. J. Med. Genet. Part A 152, 1016–1019 (2010).
- Tan, E. W. et al. Increased fracture risk and low bone mineral density in patients with loeys-dietz syndrome. Am. J. Med. Genet. A 161A, 1910–1914 (2013).
Figure 1.
Initial Magnetic resonance imaging (MRI) showing right occipital cortical-subcortical ischemic stroke (red arrows, A-B), in fluid attenuated inversion recovery (FLAIR) sequences.
Figure 1.
Initial Magnetic resonance imaging (MRI) showing right occipital cortical-subcortical ischemic stroke (red arrows, A-B), in fluid attenuated inversion recovery (FLAIR) sequences.
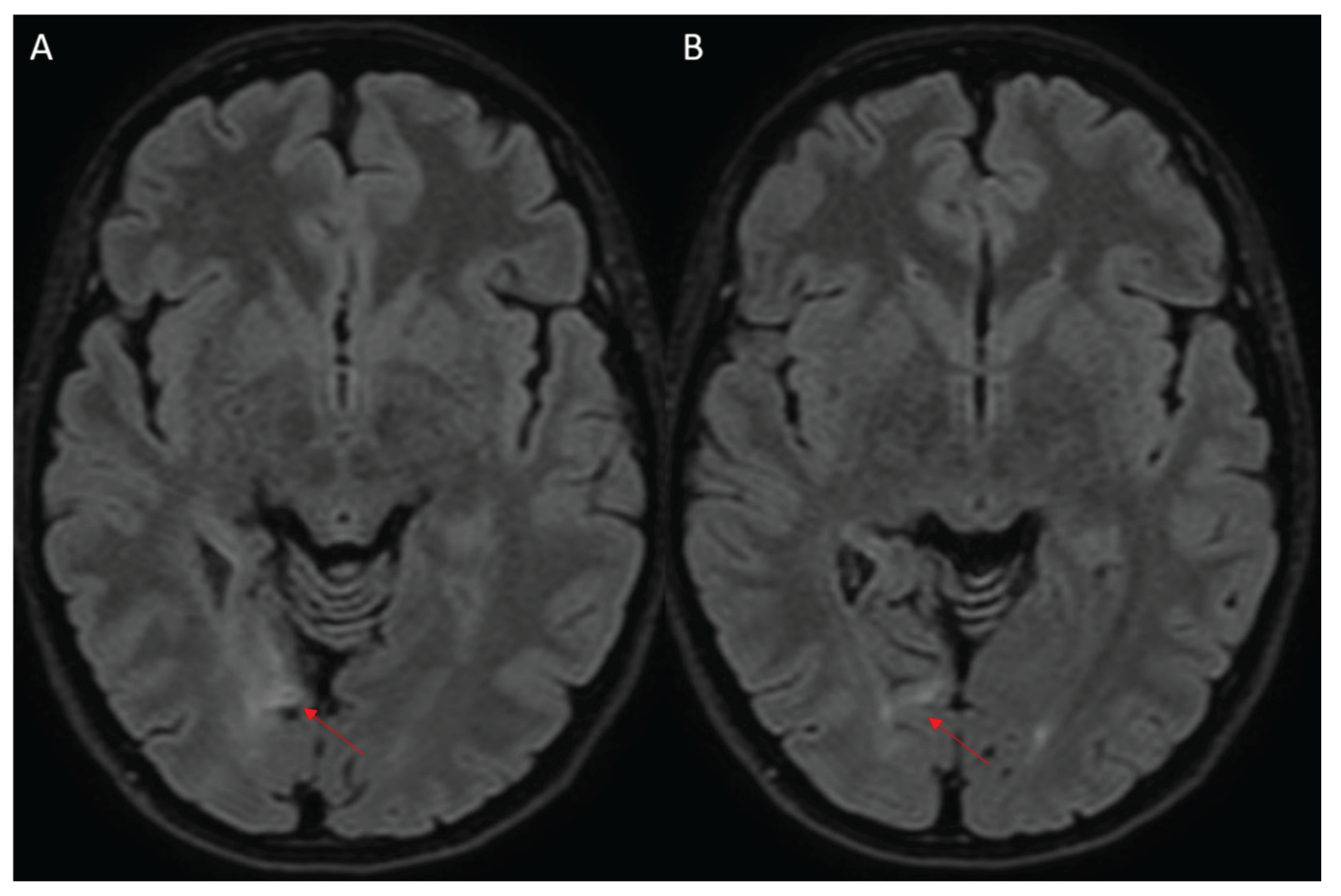
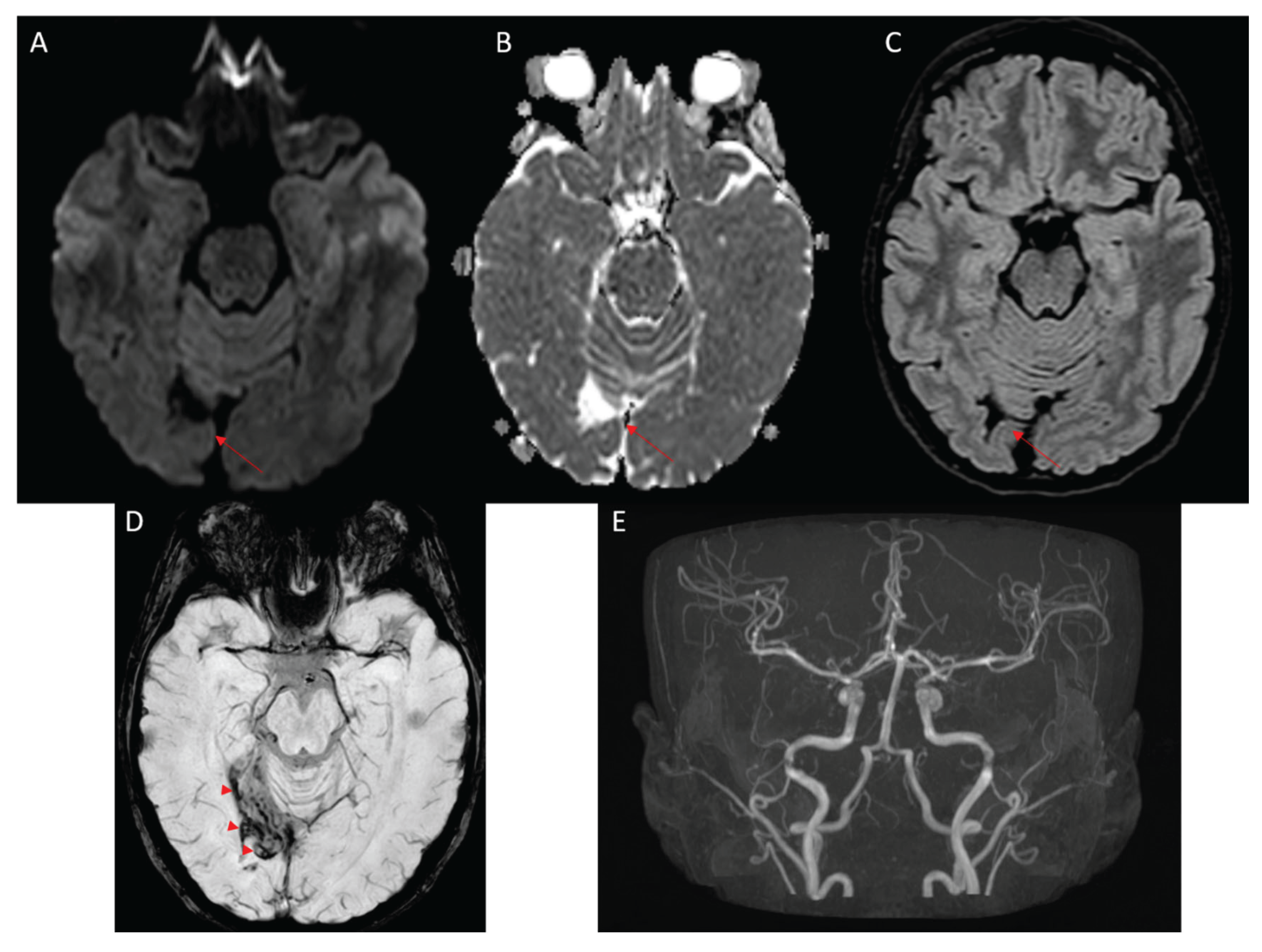
Figure 2.
Follow-up MRI showing gliotic-malacic sequelae in right occipital cortical-subcortical region (red arrows) in diffusion weighted imaging-apparent diffusion coefficient (DWI-ADC, A-B) and FLAIR (C) with hemosiderin staining (red arrowheads) in susceptibility weighted imaging (SWI, D), from focal ictal event. The TOF-3D Maximum Intensity Projection (MIP) study shows normal flow signal at the level of the main intracranial arteries, without images related to vascular malformations or aneurysms (E).
Figure 2.
Follow-up MRI showing gliotic-malacic sequelae in right occipital cortical-subcortical region (red arrows) in diffusion weighted imaging-apparent diffusion coefficient (DWI-ADC, A-B) and FLAIR (C) with hemosiderin staining (red arrowheads) in susceptibility weighted imaging (SWI, D), from focal ictal event. The TOF-3D Maximum Intensity Projection (MIP) study shows normal flow signal at the level of the main intracranial arteries, without images related to vascular malformations or aneurysms (E).
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



