Submitted:
21 April 2025
Posted:
22 April 2025
You are already at the latest version
Abstract
CB1 and CB2 cannabinoid receptors are members of the GPCR superfamily that modulate the effects of endocannabinoids. CB1 is the most abundant CB receptor in the central nervous system while CB2 is present both peripherally and in the brain. CB2 plays a role in inflammation as well as neurodegenerative and psychiatric disorders. To identify new ligands for CB2, we screened a library of FDA-approved drugs for activity at the receptor using a thallium flux assay, resulting in the discovery of the immunosuppressant mycophenolate mofetil as a potent, selective activator of CB2. Further characterization of the compound confirmed agonist activity in a variety of complementary assays including PI hydrolysis, cAMP inhibition, and β-arrestin recruitment. Radioligand binding assays established a non-competitive interaction with the site occupied by [3H]CP55,940. CB2 agonists GW-842,166X and MDA7 were also profiled, revealing that GW-842,166X exhibits a similar activity profile to mycophenolate mofetil, whereas MDA7 presents a distinct profile. These differences provide insight into the complicated CB2 pharmacology impacting preclinical and clinical studies and ultimately new treatment strategies for brain disorders.
Keywords:
cannabinoid receptor
; allosteric modulator
; mycophenolate mofetil
; GW-842
; 166X
; MDA7
; brain disorders
1. Introduction
The endocannabinoid system is comprised of two primary receptor subtypes, the cannabinoid receptor subtype 1 (CB1) and subtype 2 (CB2). While CB1 is more abundantly expressed in the brain, CB2 is expressed in the immune system and was initially thought to be a peripheral receptor [1,2]. CB1 is a potential therapeutic target for indications including nausea [3], psychosis [4], anxiety [5], and pain [6]. Subsequent studies characterized CB2 expression in the brain [7,8,9,10] and the CB2 receptor has emerged as a therapeutic target for brain disorders ranging from anxiety [11] and depression [12], to addiction [13] and schizophrenia [14,15].
A number of CB2 receptor agonists have been developed and tested in clinical trials for a variety of indications including pain and Alzheimer’s disease and several compounds have been shown to be safe for human administration. GW-842,166X is a CB2 selective agonist developed by GlaxoSmithKline [16,17] and was evaluated in Phase I trials for the indications of pain and inflammation (NCT00511524) and Phase II trials for dental pain (NCT00444769). The trials were either withdrawn or completed with no results posted. MDA7 (NTRX-07) is a CB2-preferring agonist developed by Naiguib et al. and NeuroTherapia [18,19,20]; it was progressed into Phase I trials for the indications of neuropathic pain (NCT04375436) and Alzheimer’s disease (NCT06194552) and, according to press releases, this compound is progressing into Phase II studies for Alzheimer’s disease.
As an alternative to direct CB2 activation, the use of allosteric modulators may provide an improved approach by targeting neurotransmitter-dependent activity [21]. The spatial and temporal control made possible with allosteric potentiators, along with improved subtype-selectivity, suggests that efficacy may be improved with fewer adverse side effects [22,23]. The first published small molecule positive allosteric modulator (PAM) of CB2, EC21a, has been shown to have diverse functional activity [24,25]. While reported initially as a potentiator of CB2, it has subsequently shown inverse agonist activity in a variety of independent assays; thus, a pure PAM has yet to be identified [26,27]. To discover novel compounds with CB2 PAM activity, we screened a collection of approximately 1000 FDA-approved drugs and identified mycophenolate mofetil as a putative PAM of the CB2 receptor. Additional profiling, however, revealed that this compound was actually an agonist of CB2. Mycophenolate mofetil is a widely used immunosuppressive agent [28,29,30]; this compound demonstrated CB2 activity in a variety of assays, including G Protein Inwardly Rectifying Potassium (GIRK) channel-mediated thallium flux, phosphoinositide (PI) hydrolysis, cAMP inhibition, and β-arrestin recruitment. It was also selective for CB2 versus CB1. In contrast to the parent molecule, the active form of the drug that is responsible for immunosuppression, mycophenolic acid [28,29,30], was inactive at the CB2 receptor. Radioligand binding studies indicate that mycophenolate mofetil does not interact with the binding site occupied by [3H]CP55,940 in a competitive manner. During the course of our studies, we also profiled the known CB2 agonists, GW-842,166X and MDA7. In receptor-activated GIRK assays, our studies indicated comparable agonist activity between GW-842,166X and mycophenolate mofetil but MDA7 exhibited inverse agonist activity at CB2; however, all three compounds demonstrated agonist activity in multiple signaling pathways using cAMP inhibition and β-arrestin assays. With respect to radioligand binding, GW-842,166X exhibited comparable binding activity to mycophenolate mofetil while MDA7 displayed a different binding profile. The FDA-approved immunosuppressant mycophenolate mofetil represents a novel CB2 agonist with unique properties compared to known CB2 agonists. While subtle, these differences may provide insight into the mechanism of action of CB2 modulators and prove to be significant when evaluating novel compounds as possible therapeutics for brain disorders.
2. Results
2.1. Identification of Novel Modulators of the CB2 Receptor
To identify new compounds capable of potentiating CB2 responses to the endogenous agonist 2-AG, we used a thallium flux assay measuring the ability of ligands to enhance 2-AG-induced responses in cells expressing rCB2 and GIRK channels to screen a collection of FDA-approved drugs. Compounds were added to cells loaded with thallium sensitive dye and allowed to incubate for 2.3 minutes, whereupon a submaximal concentration (EC20) of 2-AG was added to the cells and the signal was monitored for an additional 2.7 minutes. The ~1000 compound collection was initially screened in single point at a 10 µM concentration of compound. Test compounds that potentiated the cellular response to 2-AG by a minimum of 3 standard deviations above the average EC20 agonist response were selected as potential PAM hits. Sample kinetic traces of a hit from the screen, compared to EC20 and ECMax responses to 2-AG, are shown in Figure 1. 63 putative PAMs were identified; of these, 13 were confirmed as active with concentration-dependent activity at the rCB2 receptor.
2.2. Mycophenolate Mofetil is a Potent and Selective Activator of rCB2
One hit in particular, mycophenolate mofetil (Figure 2A, white circles), exhibited robust potency and efficacy at rCB2 (pEC50 = 6.64 ± 0.06, 228 nM; 74 ± 2 % Max) and was selected for additional studies. Mycophenolate mofetil is an immunosuppressant used to prevent organ transplant rejection and to treat autoimmune disorders such as lupus [28,29,30]. To determine if mycophenolate mofetil exerts its activity in the thallium flux assay through rCB2, we tested it for activity in HEK cells expressing GIRK channels but not rCB2. Submaximal concentrations of acetylcholine were used as the agonist, targeting the endogenous M4 muscarinic receptor expressed in HEK/GIRK cells [31]. Mycophenolate mofetil was found to be inactive under these conditions (Figure 2A, red circles), suggesting that its activity is dependent on the presence of the rCB2 receptor. It was also critical to determine if mycophenolate mofetil was selective for the rCB2 versus rCB1 receptors, as activation of rCB1 receptors can have negative side effects including anxiety, paranoia, and psychosis [32-34]. To determine subtype selectivity, mycophenolate mofetil was tested for activity in cells co-expressing rCB1 and GIRK channels using thallium flux. The compound did not enhance the agonist-induced response; however, mycophenolate mofetil exhibited a small inhibition of the rCB1 response to 2-AG at high concentrations. Results are shown in Figure 2A (blue circles, pEC50 < 5, -5.5 ± 1.1 % Max). Mycophenolate mofetil was also tested for activity at the human CB2 receptor and found to be active (pEC50 = 5.70 ± 0.11; 36 ± 4 % Max, data[32–34 not shown).
2.3. Mycophenolic Acid Is Inactive at CB Receptors
Mycophenolic acid is the active form of mycophenolate mofetil when used as an immunosuppressive drug or for the treatment of autoimmune disorders [35]. The morpholino ethyl ester of mycophenolic acid is a prodrug form which is rapidly metabolized to the active acid in the liver. Mycophenolic acid is a potent inhibitor of inosine-5′-monophosphate dehydrogenase [36]. To test the activity of this active metabolite, the acid was evaluated for activity at rCB2, rCB1, and parental HEK/GIRK cells using the thallium flux assay. It was found to be inactive when tested in each of these cell lines (Figure 2B), indicating mycophenolate mofetil, as opposed to the acid, is the active form of the molecule in rCB2 cells.
2.4. Mycophenolate Mofetil Has Similar Activity in the Presence and Absence of 2-AG in Thallium Flux Assays
The initial high throughput screen and secondary assays were performed in the presence of a submaximal (EC20) concentration of 2-AG to screen for PAMs, compounds that enhance the response of the receptor to an agonist. In the assay design used here, it is possible that compounds may have activity on their own in the absence of 2-AG and would instead be classified as agonists. To distinguish between these possibilities, mycophenolate mofetil was tested for activity in the presence and absence of 2-AG in cells expressing rCB2/GIRK or rCB1/GIRK using thallium flux. Increasing concentrations of mycophenolate mofetil enhanced the response of rCB2/GIRK cells in a concentration-dependent manner with similar potency and efficacy values in the absence of 2-AG (Figure 3, red circles, pEC50 = 6.81 ± 0.12, 154 nM; 71 ± 5 % Max). Comparing this activity to the PAM assay performed in Figure 2A shows that the only difference in activity between the two experiments is the elevated baseline observed in the presence of agonist due to the EC20 response of 2-AG. These results suggest that mycophenolate mofetil acts as an agonist as opposed to a PAM in rCB2/GIRK cells using thallium flux as a measure of activity. Mycophenolate mofetil decreased the response of rCB1/GIRK cells both in the presence (Figure 2A, blue circles) and absence (Figure 3, black circles, pEC50 < 5, -15 ± 3 % Max) of 2-AG to a similar value, again differentiated only by the EC20 response of 2-AG. In rCB1/GIRK cells, mycophenolate mofetil decreases the signal below baseline when added alone, although weakly, which may suggest inverse agonist activity. The decrease in signal in the presence of 30 µM mycophenolate mofetil compared to baseline is significant when added alone (p=0.002) and in the presence of 2-AG (p = 0.0003).
2.5. The Antagonist/Inverse Agonist AM630 Shifts the Mycophenolate Mofetil Concentration-Response Curve (CRC) to the Right Without Significantly Decreasing the Maximal Response
To further characterize the activity of mycophenolate mofetil, we investigated the effect of orthosteric antagonist/inverse agonist AM630 [37,38,39,40] on the concentration-response of rCB2 cells to mycophenolate mofetil. If AM630 and mycophenolate mofetil bind to the same site on the receptor, increasing concentrations of AM630 would be expected to shift the CRC to the right with no decrease in maximal response, a pattern indicative of a competitive interaction. As seen in Figure 4, AM630 had little effect on the response to mycophenolate mofetil at or below a concentration of 1 µM. At 3 and 10 µM, AM630 significantly shifted the CRC to the right compared to vehicle (pEC50 vehicle = 6.47 ± 0.16; pEC50 3 µM AM630 = 5.68 ± 0.09 (p = 0.01), pEC50 10 µM AM630 = 5.64 ± 0.16 (p = 0.02). There was no significant change in maximum signal compared to vehicle with either 3 or 10 µM AM630. While these results are not conclusive on their own, they suggest mycophenolate mofetil may have a competitive relationship with AM630.
2.6. Mycophenolate Mofetil Is Active in Phosphoinositide Hydrolysis Assays and Demonstrates Similar Activity in the Presence and Absence of 2-AG
While mycophenolate mofetil demonstrated potent agonist activity using thallium flux assays, it is important to confirm activity employing an assay that utilizes an alternate signaling pathway as CB receptors exhibit assay-dependent pharmacology [23,26,27,41,42,43,44]. In a complementary assay, we used phosphoinositide (PI) hydrolysis in the presence of the chimeric G protein, Gqi9, to assess activity. As seen in Figure 5, increasing concentrations of mycophenolate mofetil significantly increased PI hydrolysis in a concentration-dependent manner. Similar results were found in the presence (white circles, pIC50 = 6.15 ± 0.09, 700 nM; 633 ± 122 Max IP1) and absence (red circles, pIC50 = 6.28± 0.11, 521 nM; 438 ± 32 Max IP1) of 2-AG. These data are consistent with results from thallium flux assays, demonstrating mycophenolate mofetil acts as a CB2 agonist in two distinct signaling pathways.
2.7. The CB2 Receptor Agonist GW-842,166X Exhibits a Similar Functional Activity Profile Compared to Mycophenolate Mofetil
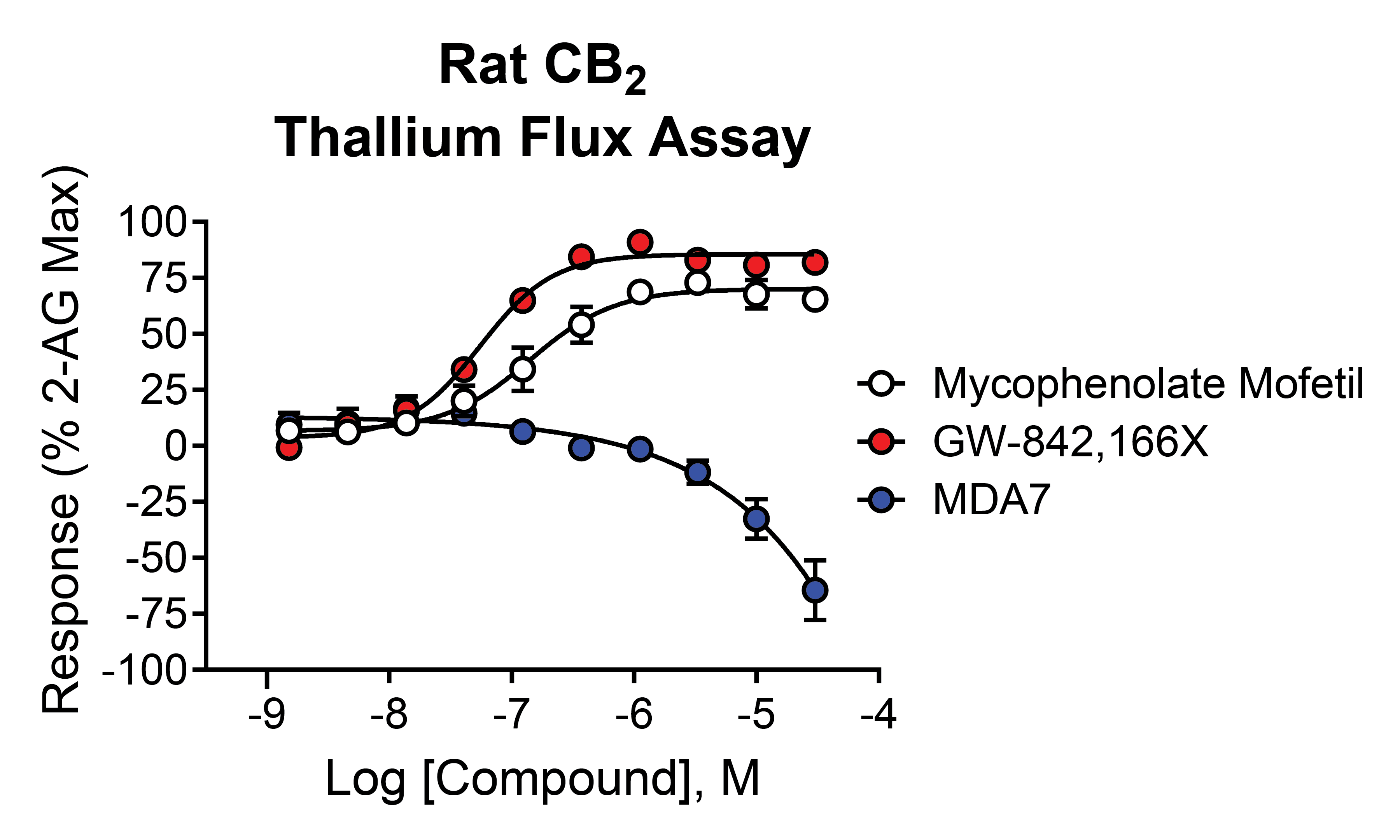
GW-842,166X is a CB2 receptor agonist that demonstrates analgesic and anti-inflammatory activity in animal models [16,45,46] and was in clinical trials for inflammation and pain [1,47]. It is structurally distinct from mycophenolate mofetil and was selected as a comparator for these studies (see Table 1 for structure). When tested for activity in rCB2/GIRK cells using thallium flux, GW-842,166X exhibited potent activity in the presence (white circles, pEC50 = 7.13 ± 0.08, 74 nM; 82 ± 4 % Max) and absence (red circles, pEC50 = 7.26 ± 0.05, 56 nM; 89 ± 5 % Max) of 2-AG as seen in Figure 6A. GW-842,166X was inactive at rCB1 in both the presence (blue circles) and absence (black circles) of 2-AG. Overall, GW-842,166X exhibits a similar functional activity profile in thallium flux assays as that seen with mycophenolate mofetil.
2.8. In Contrast to Mycophenolate Mofetil and GW-842,166X, the CB2 Receptor Agonist MDA7 Exhibits Inverse Agonist Activity at rCB2 in Thallium Flux Assays
MDA7 is a CB2 receptor modulator structurally distinct from both mycophenolate mofetil and GW-842,166X and was described as exhibiting agonist activity at CB2 in GTPγS assays [18,19,20]. Studies reported MDA7 as active in models of inflammation and neuropathic pain [20,48,49,50] and the compound was shown to induce neuroprotective effects in rodent models [51,52,53]. Currently, MDA7 is being evaluated as a novel therapeutic for the treatment of Alzheimer's disease [1,47]. To compare to the other agonists described above, we tested MDA7 for activity in both rCB2/GIRK and rCB1/GIRK cells using thallium flux. In contrast to both mycophenolate and GW-842,166X, MDA7 exhibited inhibitory activity in the presence (white circles, pEC50 < 5, -65 ± 15 Max) and absence (red circles, pEC50 < 5, -64 ± 13 Max) of 2-AG stimulation of rCB2 as seen in Figure 6B. In contrast, MDA7 activated rCB1 in the presence (blue circles, pEC50 = 6.40 ± 0.02, 395 nM; 48 ± 2 % Max) and absence (black circles, pEC50 = 6.32 ± 0.03, 477 nM; 50 ± 2 % Max) of 2-AG (Figure 6B). The detection of opposing CB2 activity in assays that target differential signaling pathways is difficult to predict but is not unanticipated, as it has been observed with other CB receptor modulators [26,27,54,55,56,57,58,59].
2.9. Mycophenolate Mofetil and Other CB2 Modulators Differentially Bind to the Site Occupied by [3H]CP55,940
Results from functional assays including thallium flux and PI hydrolysis indicate that mycophenolate mofetil acts as an agonist of rCB2 due to its ability to activate the receptor when added alone to cells expressing rCB2; however, these assays do not provide information about the binding interaction between the receptor and mycophenolate mofetil. Agonists that bind to the receptor at the site occupied by the endogenous ligand, 2-AG in this case, are considered orthosteric agonists. It is also possible for an agonist to bind and activate a receptor via a site other than the orthosteric site; these ligands are classified as allosteric agonists. To characterize the nature of the agonist activity of mycophenolate mofetil, we tested its ability to bind to the receptor in equilibrium binding assays using membranes harvested from cells expressing rCB2 using the radioligand [3H]CP55,940 [40,60]. CP55,940 has an affinity for both CB1 and CB2 receptors in the low nM range and is reported to bind to the site occupied by 2-AG [61]. The CB2 antagonist/inverse agonist AM630 fully inhibited radioligand binding with a pKi value of 7.61 ± 0.29, results consistent with those described in the literature [40]. Increasing concentrations of mycophenolate mofetil had little effect on the equilibrium binding of [3H]CP55,940 until reaching 1 µM and did not fully displace the binding when tested up to 30 µM (Figure 7, Table 1). Similar results were observed for the agonist GW-842,166X. The incomplete binding and discrepancy between the functional potency and binding affinity of these ligands suggest an overlapping or alternate binding site as opposed to a competitive interaction. In contrast, the CB2 modulator MDA7 fully inhibited radioligand binding with a pKi value of 6.60 ± 0.05. These binding results on their own suggest that MDA7 occupies the same site as [3H]CP55,940; however, the inverse agonist functional results suggest additional assays are needed to characterize the interaction.
2.10. Mycophenolate Mofetil, GW-842,166X, and MDA7 Behave as Agonists in cAMP and β-Arrestin Assays
Recent publications highlight the complex pharmacology of CB2 modulators such as EC21a and the importance of profiling CB2 modulators in assays that target different signaling pathways [26,27,43,55,59]. We have described results for thallium flux assays that measure CB receptor-mediated opening of GIRK channels and PI hydrolysis assays measuring activity via a chimeric G protein. We also evaluated the ability of mycophenolate mofetil and its comparators to modulate CB2 through two additional pathways, cAMP inhibition and β-arrestin recruitment. When added in the presence (Figure 8A) or absence (Figure 8C) of an EC20 concentration of the agonist CP55,940, mycophenolate mofetil, GW-842,166X, and MDA7 all enhanced the cAMP response to CP55,940 in cells expressing human CB2 receptors. The potency values for each (Table 2) were right-shifted compared to those observed for the thallium flux assay (Table 1). Mycophenolate mofetil also exhibited lower efficacy in cAMP assays compared to thallium flux and versus GW-842,166X and MDA7. Similarly, when tested in a β-arrestin recruitment assay in the presence (Figure 8B) or absence (Figure 8D) of an EC20 concentration of the agonist CP55,940, mycophenolate mofetil, GW-842,166X, and MDA7 also induced similar responses in the absence and presence of CP55,940 and potency values were generally right-shifted compared to thallium flux assays. MDA7 was notable in that its efficacy was lower than the other two ligands with and without 2-AG when β-arrestin recruitment was measured. These data confirm mycophenolate mofetil and comparator GW-842,166X demonstrate agonist activity across multiple assays and signaling pathways and also confirm the signal bias induced by MDA7.
3. Discussion
The endocannabinoid system, composed of CB1 and CB2 receptors, has been extensively studied as a possible therapeutic target for various neurological and inflammatory disorders. CB1 is widely expressed across brain regions, while CB2 is primarily expressed in the immune system and microglia [1,2]. Recent studies have shown CB2 expression in hippocampal, striatal, and dopaminergic neurons, suggesting the potential for CB2 modulation for the treatment of disorders including pain, addiction, and schizophrenia [1,13,14,15,47,62,63]. Reduced CB2 receptor expression and activity is correlated with an increased risk of developing schizophrenia [64,65,66,67]. We have shown that the antipsychotic effects of muscarinic M4 receptor or metabotropic glutamate receptor 1 (mGlu1) PAMs in rodent behavioral models are dependent on CB2 activation, which occurs via retrograde signaling of 2-AG [14,15]. For these reasons, it is possible that CB2 activation or potentiation may represent a novel strategy for treatment. The use of a PAM as opposed to an agonist may provide advantages, as a PAM would be predicted to produce lower levels of receptor desensitization and greater receptor specificity [22,23]. We recently profiled the first published small molecule PAM, EC21a, showing that this ligand exhibits complex pharmacology [26]. To discover novel CB2 PAMs with improved properties compared to EC21a, we conducted a screen of FDA-approved drugs, resulting in the discovery of immunosuppressant mycophenolate mofetil as a selective activator of CB2.
The initial design of the assay was intended to screen for PAMs, but subsequent assays determined mycophenolate mofetil was active in the absence of agonist. Other CB2 agonists have been or are currently being studied in preclinical and clinical trials including GW-842,166X for pain [16,45,46] and MDA7 for pain [49,50,68] and Alzheimer’s disease [51,52], but to date no CB2 agonist drugs are approved for clinical use. As shown here, both mycophenolate mofetil and GW-842,166X present clear profiles as selective agonists of CB2 in multiple assays assessing various signaling pathways including thallium flux, cAMP, and β-arrestin assays. MDA7 instead exhibits a mixed profile. In cAMP and β-arrestin assays, it acts as an agonist of the CB2 receptor; however, in thallium flux assays MDA7 demonstrates inverse agonist activity, clearly blocking the basal signal well below baseline levels. It is also not selective for CB2 in thallium flux assays as it also significantly activates the CB1 receptor. It is possible that this mixed activity profile may present challenges when assessing for potential utility as a treatment for brain disorders.
The binding data generated with [3H]CP55,940 indicate multiple binding modes/sites are present within the CB2 receptor. 2-AG is reported to share the same orthosteric binding site as CP55,940 [61] and we confirmed that 2-AG fully displaced radioligand binding (pKi = 5.46 ± 0.05). Additionally, the inverse agonist AM630 fully inhibited the equilibrium binding of the radioligand with a Ki value consistent with that reported in the literature (pKi = 7.61 ± 0.29) [40]. Mycophenolate mofetil was only able to inhibit radioligand binding to a level equal to 20% of [3H]CP55,940 total binding at 30 µM. This level of binding indicates a potential interaction with the binding site occupied by the radioligand, but the disconnect between the binding data and the functional potency generated in the same cell line (rCB2/GIRK) using thallium flux suggests the binding of mycophenolate mofetil is not completely competitive with [3H]CP55,940. It is possible the two ligands occupy the same site but have different binding contacts or that the ligands share an overlapping portion of the site. It will be important to fully characterize the binding mode of mycophenolate mofetil using mutagenesis and additional molecular pharmacology studies to more fully understand the binding and functional relationship and how it relates to future drug development. This is also the case for GW-842,166X, which exhibited a similar binding profile to that of mycophenolate mofetil with incomplete binding reaching 20% of [3H]CP55,940 total binding. The potency value of this compound in thallium flux assays (pEC50 = 7.13) revealed a left-shifted pEC50 compared to the Ki. MDA7, on the other hand, presents a different profile and fully inhibited the equilibrium binding of [3H]CP55,940 (pKi = 6.60 ± 0.05). In the rCB2/GIRK cell line measuring thallium flux, MDA7 displayed weak inverse agonist activity with a potency value significantly right-shifted in comparison (pEC50 < 5.0). The complete inhibition in the binding assay suggests a competitive interaction with the [3H]CP55,940 binding site, but the weak blockade in the functional assay indicates a disconnect between the binding affinity and the functional potency in the thallium flux assay.
In this study, we discovered that mycophenolate mofetil is a selective agonist of CB2 through screening of an FDA-approved library of drugs. Its agonist activity was confirmed in multiple assays assessing activity through different signaling pathways, including thallium flux, PI hydrolysis, cAMP inhibition, and β-arrestin recruitment. The activity profiles of the CB2 clinical candidates GW-842,166X and MDA7 were assessed and found to be similar to mycophenolate mofetil in the case of GW-842,166X but divergent for MDA7. These findings suggest that modulators of CB2 have subtle pharmacological complexities and understanding these differences may provide insight into developing better treatments for brain disorders such as schizophrenia, pain, and Alzheimer's disease.
4. Materials and Methods
4.1. Chemicals
2-Arachidonoylglycerol (2-AG) and 6-iodo-2-methyl-1-[2-(4-morpholinyl)ethyl]-1H-indol-3-yl](4-methoxyphenyl)methanone (AM630) were purchased from Cayman Chemical (Ann Arbor, MI). 2-[(1R,2R,5R)-5-hydroxy-2-(3-hydroxypropyl)cyclohexyl]-5-(2-methyloctan-2-yl)phenol (CP55,940) was purchased from Tocris Bioscience (Minneapolis, MN). [3H]-CP55,940 was obtained from Perkin Elmer (Boston, MA). 2-(2,4-dichloroanilino)-N-(oxan-4-ylmethyl)-4-(trifluoromethyl)pyrimidine-5-carboxamide (GW-842,166X) and 1-[(3-benzyl-3-methyl-2,3-dihydro-1-benzofuran-6-yl)carbonyl]piperidine (MDA7/NTRX-07) were synthesized by the Warren Center for Neuroscience Drug Discovery, Vanderbilt University, Nashville, USA as previously described [16,18,19,20].
4.2. Cell Culture
Human Embryonic Kidney (HEK) 293 cells stably expressing rCB2 and the G protein Gqi9 were maintained in Dulbecco's modified Eagle media (DMEM) containing 10% FBS, 1x antibiotic/antimycotic, 20 mM HEPES, 1 mM sodium pyruvate, 2 mM L-glutamine, 1x non-essential amino acids, 700 µg/mL G418 sulfate, and 0.6 µg/mL puromycin. HEK cells stably expressing rCB2 or rCB1 and the G protein inwardly rectifying potassium channel (GIRK) were maintained in DMEM/F12 containing 10% FBS, 1x antibiotic/antimycotic, 20 mM HEPES, 1 mM sodium pyruvate, 2 mM L-glutamine, 1x non-essential amino acids, 700 µg/mL G418 sulfate, and 0.6 µg/mL puromycin. Cells were monitored by periodic PCR detection using the LookOut Mycoplasma PCR Detection Kit (Sigma-Aldrich, St Louis, MO) to eliminate potential mycoplasma infection. Reagents were obtained from Invitrogen (Waltham, MA) unless otherwise noted.
4.3. Thallium Flux Assay
Thallium flux assays were performed as previously described in Niswender et al., 2008 [31]. HEK/GIRK cells stably expressing rCB1 or rCB2 (15,000 cells/20 µL/well) were seeded in 384-well, poly-D-lysine coated assay plates (Corning BioCoat®) (Corning Inc., Corning, NY) and incubated overnight at 37 °C in the presence of 5% CO2. The next day, medium was removed and replaced with 20 μL of 1.2 µM thallium-sensitive dye Thallos-AM (ION biosciences, San Marcos, TX), prepared as a DMSO stock solution mixed in a 1:1 ratio with 10% (w/v) pluronic acid F-127, and diluted in assay buffer (Hank's balanced salt solution, 20 mM HEPES, pH 7.4). Following 1 hour at room temperature, dye solution was removed and replaced with 20 µL assay buffer. For screening of the FDA-Approved Drug Collection, 10 mM DMSO stocks of library compounds were transferred to daughter plates using an Echo acoustic plate reformatter (Labcyte, Sunnyvale, CA) and diluted in assay buffer to a 2x final concentration. For concentration-response curve experiments, compounds were serially diluted 1:3 into 10-point concentration response curves in DMSO, transferred to daughter plates using the Echo or Bravo Automated Liquid Handling Platform (Agilent Technologies, Santa Clara, CA), and diluted in assay buffer to a 2x final concentration. Thallium flux was measured using the Hamamatsu FDSS/μCELL Kinetic Plate Imager (Bridgewater, NJ). After establishment of a fluorescence baseline (excitation, 480 ± 20 nm; emission, 540 ± 30 nm), 20 µL (2x) of test compound was added to the cells at 1 second and the response was measured. 140 seconds later, 10 µL (5x) of an EC20 concentration of agonist (PAM mode) or vehicle (AGO mode) in thallium stimulus buffer (125 mM NaHCO3, 1.8 mM CaSO4, 1 mM MgSO4, 5 mM glucose, 12 mM Tl2SO4, 10 mM HEPES, 0.5% BSA, pH 7.4) was added to the cells, and the response of the cells was measured for an additional 159 seconds (data were acquired for 300 seconds total at 0.5 Hz for 140 seconds and 1 Hz for 160 seconds). Raw kinetic data were analyzed in a multi-step process. 1) Fluorescence readings for each time point in a well were divided by the fluorescence reading at the initial time point to account for differences in cell number, non-uniform illumination, and dye-loading. 2) The slope value for each kinetic trace was calculated for the time window of 145-155 seconds, a window occurring directly after the second addition. 3) The average slope was calculated for wells containing vehicle and this value was subtracted from all wells. 4) Vehicle-subtracted slope was normalized to the relevant maximal agonist signal for each assay. For concentration response curves, normalized data were fit to a four-parameter logistic equation using GraphPad Prism (La Jolla, CA) or the Dotmatics software platform (Dotmatics, Bishop’s Stortford, UK).
where A is the molar concentration of the compound; bottom and top denote the lower and upper plateaus of the concentration-response curve; Hillslope is the Hill coefficient that describes the steepness of the curve; and EC50 is the molar concentration of compound required to generate a response halfway between the top and bottom. Data shown represent the mean ± standard error of the pEC50 or maximal response. Experiments were performed in duplicate or triplicate and repeated a minimum of three separate times.
4.4. PI Hydrolysis Assay
One day prior to experimentation, selected HEK/Gqi9 monoclonal cells stably expressing rat CB2 were plated onto poly-D-lysine coated clear-bottom 384-well plates (15,000 cells per well) in DMEM supplemented with 10% FBS and 20 mM HEPES. Ten minutes prior to the assay, cell culture medium was replaced with 20 µL 37 °C Hank's balanced salt solution (HBSS, with Ca2+, Mg2+, glucose). IP1 stimulation was then initiated by adding 5 µL of 5x agonist dissolved in HBSS plus 40 mM Li+, and cells were incubated for an additional hour before aspiration and addition of Lysis Buffer. IP1 levels were determined using the Cisbio HTRF IP-ONE assay kit per the manufacturer’s instructions, and fluorescence was measured using an Envision plate reader (PerkinElmer, Waltham, MA). Data were acquired as HTRF ratio (665/620) and expressed as nanomolar levels of IP1.
4.5. Radioligand Binding Assays
Membranes were made from HEK/GIRK cells stably expressing rat CB2. Radioligand competition binding assays were performed as previously described with minor modifications. In brief, compounds were serially diluted into assay buffer with 0.1% bovine serum albumin (BSA) and added to each well of a 96-well plate, along with 20 μg/well cell membrane and approximately 500 pM [3H]-CP55,940 (specific activity = 104 Ci/mmol, PerkinElmer, Waltham, MA). Following a 3-hour incubation period on shaker at room temperature, the membrane-bound ligand was separated from free ligand by filtration through glass fiber 96-well filter plates (Unifilter-96, GF/B; PerkinElmer, Waltham, MA). Forty microliters of scintillation fluid were added to each well, and the membrane-bound radioactivity was determined by scintillation counting (Microbeta2; Revvity, Waltham, MA). Nonspecific binding was determined using 10 μM of cold CP55,940.
4.6. cAMP Assays
Assays were performed by Eurofins Discovery (Freemont, CA). cAMP Hunter cell lines were expanded from freezer stocks according to standard procedures. Cells were seeded in a total volume of 20 μL into white walled, 384-well microplates and incubated at 37 °C for the appropriate time prior to testing. cAMP modulation was determined using the DiscoverX HitHunter cAMP XS+ assay. For agonist determination, cells were incubated with sample in the presence of EC80 forskolin to induce response. Media was aspirated from cells and replaced with 10 μL HBSS/10mM HEPES. Intermediate dilution of sample stocks was performed to generate 4X sample in assay buffer and 5 μL of 4X sample was added to cells. 5 μL of 4X EC80 forskolin diluted in HBSS/HEPES was added and incubated at 37 °C for 30 minutes. Final assay vehicle concentration was 1%. For allosteric determination, cells were pre-incubated with sample followed by agonist induction at the EC20 concentration. Media was aspirated from cells and replaced with 10 μL HBSS/10mM HEPES. Intermediate dilution of sample stocks was performed to generate 4X sample in assay buffer. 5 μL of 4X compound was added to the cells and incubated at room temperature or 37 °C for 5 minutes. 5 μL of 4X EC20 agonist was added to the cells and incubated at 37 °C for 30 minutes. EC80 forskolin was included.
After appropriate compound incubation, assay signal was generated through incubation with 5μL cAMP XS+ Ab reagent followed by 20 μL cAMP XS+ ED/CL lysis cocktail for one hour at room temperature. 20 μL cAMP XS+ EA reagent for two hours at room temperature. Microplates were read following signal generation with a PerkinElmer EnvisionTM instrument for chemiluminescent signal detection.
Compound activity was analyzed using CBIS data analysis suite (ChemInnovation, CA). For agonist mode assays, percentage activity is calculated using the following formula: % Activity = 100% x (1 - (mean RLU of test sample - mean RLU of MAX control) / (mean RLU of vehicle control - mean RLU of MAX control)).
For Gi positive allosteric mode assays, percentage modulation was calculated using the following formula: % Modulation =100% x (1-(mean RLU of test sample - mean RLU of MAX control) / (mean RLU of EC20 control - mean RLU of MAX control)).
4.7.β-. Arrestin Assays
Assays were performed by Eurofins Discovery (Freemont, CA). PathHunter cell lines were expanded from freezer stocks according to standard procedures. Cells were seeded in a total volume of 20 μL into white walled, 384-well microplates and incubated at 37 °C for the appropriate time prior to testing. For agonist determination, cells were incubated with sample to induce response. Intermediate dilution of sample stocks was performed to generate 5X sample in assay buffer. 5 μL of 5X sample was added to cells and incubated at 37 °C or room temperature for at least 120 minutes. Final assay vehicle concentration was 1%. For allosteric modulator determination, cells were pre-incubated with sample followed by agonist challenge at the EC20. Intermediate dilution of sample stocks was performed to generate 5X sample in assay buffer. 5 μL of 5X sample was added to cells and incubated at 37 °C or room temperature for 15 minutes. Vehicle concentration was 1%. 5 μL of 6X EC20 agonist in assay buffer was added to the cells and incubated at 37 °C or room temperature for at least 120 minutes.
Assay signal was generated through a single addition of 15 μL (50% v/v) of PathHunter Detection reagent cocktail, followed by a one-hour incubation at room temperature. Microplates were read following signal generation with a PerkinElmer EnvisionTM instrument for chemiluminescent signal detection.
Compound activity was analyzed using CBIS data analysis suite (ChemInnovation, CA). For agonist mode assays, percentage activity was calculated using the following formula: % Activity = 100% x (mean RLU of test sample — mean RLU of vehicle control) / (mean MAX control ligand — mean RLU of vehicle control).
For positive allosteric mode assays, percentage modulation was calculated using the following formula: % Modulation = 100% x ((mean RLU of test sample — mean RLU of EC20 control) / (mean RLU of MAX control ligand — mean RLU of EC20 control)).
Author Contributions
Conceptualization, C.M.N.; methodology, A.L.R., A.Q., and C.M.N; formal analysis, A.L.R., A.Q., and C.M.N; investigation, A.Q., A.H., H.E.K., M.Q., (pharmacology) A.M.B., L.B., and D. W. (chemistry); resources, C.M.N. and C.W.L.; writing—original draft preparation, A.L.R.; writing—review and editing, A.L.R., C.M.N., A.Q., A.H., H.E.K., M.Q., C.W.L., A.M.B., L.B., and D. W. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by National Institutes of Health under Grant Numbers MH119673 and MH062464, research funds from the Warren Center for Neuroscience Drug Discovery, and a generous gift from the William K. Warren Foundation.
Institutional Review Board Statement
Not applicable.
Data Availability Statement
The authors declare that all processed data supporting the findings of this study are available within the paper. Raw data are available on request from the corresponding author.
Acknowledgments
We thank the William K. Warren Foundation for their funding toward the Warren Center for Neuroscience Drug Discovery.
Conflicts of Interest
The authors declare no conflicts of interest.
Abbreviations
The following abbreviations are used in this manuscript:
| 2-AG | 2-Arachidonylglycerol |
| AM630 | 6-Iodo-2-methyl-1-[2-(4-morpholinyl)ethyl]-1H-indol-3-yl](4-methoxyphenyl)methanone |
| cAMP | cyclic adenosine monophosphate |
| BSA | bovine serum albumin |
| CB1 | cannabinoid receptor subtype 1 |
| CB2 | cannabinoid receptor subtype 2 |
| CP55,940 | 2-[(1R,2R,5R)-5-hydroxy-2-(3-hydroxypropyl)cyclohexyl]-5-(2-methyloctan-2-yl)phenol |
| CRC | concentration response curve |
| DMEM | Dulbecco's Modified Eagle Media |
| DMSO | dimethyl sulfoxide |
| EC21a | N-(5-bromo-1-(4-fluorobenzyl)-4-methyl-2-oxo-1,2-dihydropyridin-3-yl)cycloheptanecarboxamide |
| F12 | Ham's F-12 Nutrient Mixture |
| FBS | fetal bovine serum |
| FDA | Food and Drug Administration |
| GPCR | G protein-coupled receptor |
| GIRK | G protein-regulated inwardly rectifying potassium channel |
| GW-842,166X | 2-(2,4-dichloroanilino)-N-(oxan-4-ylmethyl)-4-(trifluoromethyl)pyrimidine-5-carboxamide |
| HBSS | Hank's balanced salt solution |
| HEK | human embryonic kidney |
| HEPES | 4-(2-hydroxyethyl)piperazine-1-ethanesulfonic acid |
| IP1 | inositol monophosphate |
| MDA7 | 1-[(3-benzyl-3-methyl-2,3-dihydro-1-benzofuran-6-yl)carbonyl]piperidine |
| mGlu1 | metabotropic glutamate receptor subtype 1 |
| M4 | muscarinic acetylcholine receptor subtype 4 |
| PAM | positive allosteric modulator |
| PI | phosphoinositide |
| SEM | standard error of the mean |
References
- García-Gutiérrez, M.S.; Torregrosa, A.B.; Navarrete, F.; Navarro, D.; Manzanares, J. A comprehensive review of the multifaceted role of cannabinoid receptor type 2 in neurodegenerative and neuropsychiatric disorders. Pharmacol Res 2025, 213, 107657. [Google Scholar] [CrossRef]
- Howlett, A.C.; Barth, F.; Bonner, T.I.; Cabral, G.; Casellas, P.; Devane, W.A.; Felder, C.C.; Herkenham, M.; Mackie, K.; Martin, B.R.; et al. International Union of Pharmacology. XXVII. Classification of cannabinoid receptors. Pharmacol Rev 2002, 54, 161–202. [Google Scholar] [CrossRef] [PubMed]
- Elgohary, R.; Salama, A.; Omara, E.A. Protective Effects of Cannabis sativa on chemotherapy-induced nausea in a rat: Involvement of CB1 receptors. Fundam Clin Pharmacol 2023, 37, 137–146. [Google Scholar] [CrossRef]
- Hill, M.N.; Haney, M.; Hillard, C.J.; Karhson, D.S.; Vecchiarelli, H.A. The endocannabinoid system as a putative target for the development of novel drugs for the treatment of psychiatric illnesses. Psychol Med 2023, 53, 7006–7024. [Google Scholar] [CrossRef] [PubMed]
- Haller, J.; Varga, B.; Ledent, C.; Freund, T.F. CB1 cannabinoid receptors mediate anxiolytic effects: convergent genetic and pharmacological evidence with CB1-specific agents. Behav Pharmacol 2004, 15, 299–304. [Google Scholar] [CrossRef]
- Quintero, J.M.; Diaz, L.E.; Galve-Roperh, I.; Bustos, R.H.; Leon, M.X.; Beltran, S.; Dodd, S. The endocannabinoid system as a therapeutic target in neuropathic pain: a review. Expert Opin Ther Targets 2024, 28, 739–755. [Google Scholar] [CrossRef]
- Van Sickle, M.D.; Duncan, M.; Kingsley, P.J.; Mouihate, A.; Urbani, P.; Mackie, K.; Stella, N.; Makriyannis, A.; Piomelli, D.; Davison, J.S.; et al. Identification and functional characterization of brainstem cannabinoid CB2 receptors. Science 2005, 310, 329–332. [Google Scholar] [CrossRef] [PubMed]
- Gong, J.P.; Onaivi, E.S.; Ishiguro, H.; Liu, Q.R.; Tagliaferro, P.A.; Brusco, A.; Uhl, G.R. Cannabinoid CB2 receptors: immunohistochemical localization in rat brain. Brain Res 2006, 1071, 10–23. [Google Scholar] [CrossRef]
- Onaivi, E.S.; Ishiguro, H.; Gong, J.P.; Patel, S.; Perchuk, A.; Meozzi, P.A.; Myers, L.; Mora, Z.; Tagliaferro, P.; Gardner, E.; et al. Discovery of the presence and functional expression of cannabinoid CB2 receptors in brain. Ann N Y Acad Sci 2006, 1074, 514–536. [Google Scholar] [CrossRef]
- García-Gutiérrez, M.S.; Pérez-Ortiz, J.M.; Gutiérrez-Adán, A.; Manzanares, J. Depression-resistant endophenotype in mice overexpressing cannabinoid CB(2) receptors. Br J Pharmacol 2010, 160, 1773–1784. [Google Scholar] [CrossRef]
- García-Gutiérrez, M.S.; Manzanares, J. Overexpression of CB2 cannabinoid receptors decreased vulnerability to anxiety and impaired anxiolytic action of alprazolam in mice. J Psychopharmacol 2011, 25, 111–120. [Google Scholar] [CrossRef] [PubMed]
- Onaivi, E.S.; Ishiguro, H.; Sejal, P.; Meozzi, P.A.; Myers, L.; Tagliaferro, P.; Hope, B.; Leonard, C.M.; Uhl, G.R.; Brusco, A.; et al. Methods to study the behavioral effects and expression of CB2 cannabinoid receptor and its gene transcripts in the chronic mild stress model of depression. Methods Mol Med 2006, 123, 291–298. [Google Scholar] [CrossRef]
- Ishiguro, H.; Iwasaki, S.; Teasenfitz, L.; Higuchi, S.; Horiuchi, Y.; Saito, T.; Arinami, T.; Onaivi, E.S. Involvement of cannabinoid CB2 receptor in alcohol preference in mice and alcoholism in humans. Pharmacogenomics J 2007, 7, 380–385. [Google Scholar] [CrossRef]
- Foster, D.J.; Wilson, J.M.; Remke, D.H.; Mahmood, M.S.; Uddin, M.J.; Wess, J.; Patel, S.; Marnett, L.J.; Niswender, C.M.; Jones, C.K.; et al. Antipsychotic-like Effects of M4 Positive Allosteric Modulators Are Mediated by CB2 Receptor-Dependent Inhibition of Dopamine Release. Neuron 2016, 91, 1244–1252. [Google Scholar] [CrossRef] [PubMed]
- Yohn, S.E.; Foster, D.J.; Covey, D.P.; Moehle, M.S.; Galbraith, J.; Garcia-Barrantes, P.M.; Cho, H.P.; Bubser, M.; Blobaum, A.L.; Joffe, M.E.; et al. Activation of the mGlu1 metabotropic glutamate receptor has antipsychotic-like effects and is required for efficacy of M4 muscarinic receptor allosteric modulators. Mol Psychiatry 2020, 25, 2786–2799. [Google Scholar] [CrossRef]
- Giblin, G.M.; O'Shaughnessy, C.T.; Naylor, A.; Mitchell, W.L.; Eatherton, A.J.; Slingsby, B.P.; Rawlings, D.A.; Goldsmith, P.; Brown, A.J.; Haslam, C.P.; et al. Discovery of 2-[(2,4-dichlorophenyl)amino]-N-[(tetrahydro- 2H-pyran-4-yl)methyl]-4-(trifluoromethyl)- 5-pyrimidinecarboxamide, a selective CB2 receptor agonist for the treatment of inflammatory pain. J Med Chem 2007, 50, 2597–2600. [Google Scholar] [CrossRef] [PubMed]
- Ostenfeld, T.; Price, J.; Albanese, M.; Bullman, J.; Guillard, F.; Meyer, I.; Leeson, R.; Costantin, C.; Ziviani, L.; Nocini, P.F.; et al. A randomized, controlled study to investigate the analgesic efficacy of single doses of the cannabinoid receptor-2 agonist GW842166, ibuprofen or placebo in patients with acute pain following third molar tooth extraction. Clin J Pain 2011, 27, 668–676. [Google Scholar] [CrossRef]
- Attala, M.N.; Brown, D.L. Neuroprotective CB2 Receptor Agonists. WO 2014/011949 A2, January 16, 2014.
- Attala, M.N.; Diaz, P. Heterocycle Modulators of Cannabinoid Receptors. US 9,339,486 B2, May 17, 2016.
- Naguib, M.; Diaz, P.; Xu, J.J.; Astruc-Diaz, F.; Craig, S.; Vivas-Mejia, P.; Brown, D.L. MDA7: a novel selective agonist for CB2 receptors that prevents allodynia in rat neuropathic pain models. Br J Pharmacol 2008, 155, 1104–1116. [Google Scholar] [CrossRef]
- Wootten, D.; Christopoulos, A.; Sexton, P.M. Emerging paradigms in GPCR allostery: implications for drug discovery. Nat Rev Drug Discov 2013, 12, 630–644. [Google Scholar] [CrossRef]
- Lindsley, C.W.; Emmitte, K.A.; Hopkins, C.R.; Bridges, T.M.; Gregory, K.J.; Niswender, C.M.; Conn, P.J. Practical Strategies and Concepts in GPCR Allosteric Modulator Discovery: Recent Advances with Metabotropic Glutamate Receptors. Chem Rev 2016, 116, 6707–6741. [Google Scholar] [CrossRef]
- Gado, F.; Meini, S.; Bertini, S.; Digiacomo, M.; Macchia, M.; Manera, C. Allosteric modulators targeting cannabinoid cb1 and cb2 receptors: implications for drug discovery. Future Med Chem 2019, 11, 2019–2037. [Google Scholar] [CrossRef] [PubMed]
- Gado, F.; Di Cesare Mannelli, L.; Lucarini, E.; Bertini, S.; Cappelli, E.; Digiacomo, M.; Stevenson, L.A.; Macchia, M.; Tuccinardi, T.; Ghelardini, C.; et al. Identification of the First Synthetic Allosteric Modulator of the CB2 Receptors and Evidence of Its Efficacy for Neuropathic Pain Relief. J Med Chem 2019, 62, 276–287. [Google Scholar] [CrossRef]
- Shapiro, L.; Gado, F.; Manera, C.; Escayg, A. Allosteric modulation of the cannabinoid 2 receptor confers seizure resistance in mice. Neuropharmacology 2021, 188, 108448. [Google Scholar] [CrossRef] [PubMed]
- Qi, A.; Han, X.; Quitalig, M.; Wu, J.; Christov, P.P.; Jeon, K.; Jana, S.; Kim, K.; Engers, D.W.; Lindsley, C.W.; et al. The cannabinoid CB2 receptor positive allosteric modulator EC21a exhibits complicated pharmacology in vitro. J Recept Signal Transduct Res 2024, 44, 151–159. [Google Scholar] [CrossRef] [PubMed]
- Farooq, Z.; Delre, P.; Iliadis, S.; Mangiatordi, G.F.; Contino, M.; Howell, L.A.; McCormick, P.J. Identification of a Cannabinoid Receptor 2 Allosteric Site Using Computational Modeling and Pharmacological Analysis. ACS Pharmacology & Translational Science 2025, 8, 423–434. [Google Scholar] [CrossRef]
- Allison, A.C.; Eugui, E.M. Immunosuppressive and other anti-rheumatic activities of mycophenolate mofetil. Agents Actions Suppl 1993, 44, 165–188. [Google Scholar]
- Eugui, E.M.; Allison, A.C. Immunosuppressive activity of mycophenolate mofetil. Ann N Y Acad Sci 1993, 685, 309–329. [Google Scholar] [CrossRef]
- Ritter, M.L.; Pirofski, L. Mycophenolate mofetil: effects on cellular immune subsets, infectious complications, and antimicrobial activity. Transpl Infect Dis 2009, 11, 290–297. [Google Scholar] [CrossRef]
- Niswender, C.M.; Johnson, K.A.; Luo, Q.; Ayala, J.E.; Kim, C.; Conn, P.J.; Weaver, C.D. A novel assay of Gi/o-linked G protein-coupled receptor coupling to potassium channels provides new insights into the pharmacology of the group III metabotropic glutamate receptors. Mol Pharmacol 2008, 73, 1213–1224. [Google Scholar] [CrossRef]
- Martin, R.S.; Secchi, R.L.; Sung, E.; Lemaire, M.; Bonhaus, D.W.; Hedley, L.R.; Lowe, D.A. Effects of cannabinoid receptor ligands on psychosis-relevant behavior models in the rat. Psychopharmacology (Berl) 2003, 165, 128–135. [Google Scholar] [CrossRef]
- Gobira, P.H.; LaMar, J.; Marques, J.; Sartim, A.; Silveira, K.; Santos, L.; Wegener, G.; Guimaraes, F.S.; Mackie, K.; Lu, H.C.; et al. CB1 Receptor Silencing Attenuates Ketamine-Induced Hyperlocomotion Without Compromising Its Antidepressant-Like Effects. Cannabis Cannabinoid Res 2023, 8, 768–778. [Google Scholar] [CrossRef] [PubMed]
- Leo, L.M.; Abood, M.E. CB1 Cannabinoid Receptor Signaling and Biased Signaling. Molecules 2021, 26. [Google Scholar] [CrossRef]
- Allison, A.C.; Eugui, E.M. Immunosuppressive and other effects of mycophenolic acid and an ester prodrug, mycophenolate mofetil. Immunol Rev 1993, 136, 5–28. [Google Scholar] [CrossRef]
- Allison, A.C.; Kowalski, W.J.; Muller, C.D.; Eugui, E.M. Mechanisms of action of mycophenolic acid. Ann N Y Acad Sci 1993, 696, 63–87. [Google Scholar] [CrossRef] [PubMed]
- Pertwee, R.; Griffin, G.; Fernando, S.; Li, X.; Hill, A.; Makriyannis, A. AM630, a competitive cannabinoid receptor antagonist. Life Sci 1995, 56, 1949–1955. [Google Scholar] [CrossRef]
- Hosohata, K.; Quock, R.M.; Hosohata, Y.; Burkey, T.H.; Makriyannis, A.; Consroe, P.; Roeske, W.R.; Yamamura, H.I. AM630 is a competitive cannabinoid receptor antagonist in the guinea pig brain. Life Sci 1997, 61, PL115–PL118. [Google Scholar] [CrossRef]
- Hosohata, Y.; Quock, R.M.; Hosohata, K.; Makriyannis, A.; Consroe, P.; Roeske, W.R.; Yamamura, H.I. AM630 antagonism of cannabinoid-stimulated [35S]GTP gamma S binding in the mouse brain. Eur J Pharmacol 1997, 321, R1–R3. [Google Scholar] [CrossRef] [PubMed]
- Ross, R.A.; Brockie, H.C.; Stevenson, L.A.; Murphy, V.L.; Templeton, F.; Makriyannis, A.; Pertwee, R.G. Agonist-inverse agonist characterization at CB1 and CB2 cannabinoid receptors of L759633, L759656, and AM630. Br J Pharmacol 1999, 126, 665–672. [Google Scholar] [CrossRef] [PubMed]
- Navarro, G.; Reyes-Resina, I.; Rivas-Santisteban, R.; Sánchez de Medina, V.; Morales, P.; Casano, S.; Ferreiro-Vera, C.; Lillo, A.; Aguinaga, D.; Jagerovic, N.; et al. Cannabidiol skews biased agonism at cannabinoid CB1 and CB2 receptors with smaller effect in CB1-CB2 heteroreceptor complexes. Biochem Pharmacol 2018, 157, 148–158. [Google Scholar] [CrossRef]
- Thomas, A.; Baillie, G.L.; Phillips, A.M.; Razdan, R.K.; Ross, R.A.; Pertwee, R.G. Cannabidiol displays unexpectedly high potency as an antagonist of CB1 and CB2 receptor agonists in vitro. Br J Pharmacol 2007, 150, 613–623. [Google Scholar] [CrossRef]
- Gado, F.; Ferrisi, R.; Polini, B.; Mohamed, K.A.; Ricardi, C.; Lucarini, E.; Carpi, S.; Domenichini, F.; Stevenson, L.A.; Rapposelli, S.; et al. Design, Synthesis, and Biological Activity of New CB2 Receptor Ligands: from Orthosteric and Allosteric Modulators to Dualsteric/Bitopic Ligands. J Med Chem 2022, 65, 9918–9938. [Google Scholar] [CrossRef]
- Ferrisi, R.; Polini, B.; Ricardi, C.; Gado, F.; Mohamed, K.A.; Baron, G.; Faiella, S.; Poli, G.; Rapposelli, S.; Saccomanni, G.; et al. New Insights into Bitopic Orthosteric/Allosteric Ligands of Cannabinoid Receptor Type 2. Int J Mol Sci 2023, 24. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.R.; Tu, J.X.; Qiao, A.Q.; Chen, L.J. GW842166X Alleviates Osteoarthritis by Repressing LPS-mediated Chondrocyte Catabolism in Mice. Curr Med Sci 2022, 42, 1046–1054. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Yu, H.; Chen, B.; Friedman, V.; Mu, L.; Kelly, T.J.; Ruiz-Pérez, G.; Zhao, L.; Bai, X.; Hillard, C.J.; et al. CB2 Agonist GW842166x Protected against 6-OHDA-Induced Anxiogenic- and Depressive-Related Behaviors in Mice. Biomedicines 2022, 10. [Google Scholar] [CrossRef] [PubMed]
- Naikoo, R.A.; Painuli, R.; Akhter, Z.; Singh, P.P. Cannabinoid receptor 2 (CB2) modulators: A patent review (2016-2024). Bioorg Chem 2024, 153, 107775. [Google Scholar] [CrossRef]
- Diaz, P.; Phatak, S.S.; Xu, J.; Fronczek, F.R.; Astruc-Diaz, F.; Thompson, C.M.; Cavasotto, C.N.; Naguib, M. 2,3-Dihydro-1-benzofuran derivatives as a series of potent selective cannabinoid receptor 2 agonists: design, synthesis, and binding mode prediction through ligand-steered modeling. ChemMedChem 2009, 4, 1615–1629. [Google Scholar] [CrossRef]
- Xu, J.J.; Diaz, P.; Bie, B.; Astruc-Diaz, F.; Wu, J.; Yang, H.; Brown, D.L.; Naguib, M. Spinal gene expression profiling and pathways analysis of a CB2 agonist (MDA7)-targeted prevention of paclitaxel-induced neuropathy. Neuroscience 2014, 260, 185–194. [Google Scholar] [CrossRef]
- Wu, J.; Hocevar, M.; Bie, B.; Foss, J.F.; Naguib, M. Cannabinoid Type 2 Receptor System Modulates Paclitaxel-Induced Microglial Dysregulation and Central Sensitization in Rats. J Pain 2019, 20, 501–514. [Google Scholar] [CrossRef]
- Wu, J.; Bie, B.; Yang, H.; Xu, J.J.; Brown, D.L.; Naguib, M. Activation of the CB2 receptor system reverses amyloid-induced memory deficiency. Neurobiol Aging 2013, 34, 791–804. [Google Scholar] [CrossRef]
- Wu, J.; Hocevar, M.; Foss, J.F.; Bie, B.; Naguib, M. Activation of CB2 receptor system restores cognitive capacity and hippocampal Sox2 expression in a transgenic mouse model of Alzheimer's disease. Eur J Pharmacol 2017, 811, 12–20. [Google Scholar] [CrossRef]
- Xu, J.; Tang, Y.; Xie, M.; Bie, B.; Wu, J.; Yang, H.; Foss, J.F.; Yang, B.; Rosenquist, R.W.; Naguib, M. Activation of cannabinoid receptor 2 attenuates mechanical allodynia and neuroinflammatory responses in a chronic post-ischemic pain model of complex regional pain syndrome type I in rats. Eur J Neurosci 2016, 44, 3046–3055. [Google Scholar] [CrossRef]
- Demuth, D.G.; Molleman, A. Cannabinoid signalling. Life Sci 2006, 78, 549–563. [Google Scholar] [CrossRef]
- Soethoudt, M.; Grether, U.; Fingerle, J.; Grim, T.W.; Fezza, F.; de Petrocellis, L.; Ullmer, C.; Rothenhäusler, B.; Perret, C.; van Gils, N.; et al. Cannabinoid CB2 receptor ligand profiling reveals biased signalling and off-target activity. Nat Commun 2017, 8, 13958. [Google Scholar] [CrossRef] [PubMed]
- Howlett, A.C.; Abood, M.E. CB1 and CB2 Receptor Pharmacology. Adv Pharmacol 2017, 80, 169–206. [Google Scholar] [CrossRef]
- Gasperi, V.; Guzzo, T.; Topai, A.; Gambacorta, N.; Ciriaco, F.; Nicolotti, O.; Maccarrone, M. Recent Advances on Type-2 Cannabinoid (CB2) Receptor Agonists and their Therapeutic Potential. Curr Med Chem 2023, 30, 1420–1457. [Google Scholar] [CrossRef] [PubMed]
- Legare, C.A.; Raup-Konsavage, W.M.; Vrana, K.E. Therapeutic Potential of Cannabis, Cannabidiol, and Cannabinoid-Based Pharmaceuticals. Pharmacology 2022, 107, 131–149. [Google Scholar] [CrossRef]
- Zagzoog, A.; Brandt, A.L.; Black, T.; Kim, E.D.; Burkart, R.; Patel, M.; Jin, Z.; Nikolaeva, M.; Laprairie, R.B. Assessment of select synthetic cannabinoid receptor agonist bias and selectivity between the type 1 and type 2 cannabinoid receptor. Sci Rep 2021, 11, 10611. [Google Scholar] [CrossRef] [PubMed]
- Sugiura, T.; Kondo, S.; Sukagawa, A.; Nakane, S.; Shinoda, A.; Itoh, K.; Yamashita, A.; Waku, K. 2-Arachidonoylglycerol: a possible endogenous cannabinoid receptor ligand in brain. Biochem Biophys Res Commun 1995, 215, 89–97. [Google Scholar] [CrossRef]
- Yuan, J.; Yang, B.; Hou, G.; Xie, X.Q.; Feng, Z. Targeting the endocannabinoid system: Structural determinants and molecular mechanism of allosteric modulation. Drug Discov Today 2023, 28, 103615. [Google Scholar] [CrossRef]
- Aghazadeh Tabrizi, M.; Baraldi, P.G.; Borea, P.A.; Varani, K. Medicinal Chemistry, Pharmacology, and Potential Therapeutic Benefits of Cannabinoid CB2 Receptor Agonists. Chem Rev 2016, 116, 519–560. [Google Scholar] [CrossRef]
- Nevalainen, T. Recent development of CB2 selective and peripheral CB1/CB2 cannabinoid receptor ligands. Curr Med Chem 2014, 21, 187–203. [Google Scholar] [CrossRef] [PubMed]
- Ishiguro, H.; Kibret, B.G.; Horiuchi, Y.; Onaivi, E.S. Potential Role of Cannabinoid Type 2 Receptors in Neuropsychiatric and Neurodegenerative Disorders. Front Psychiatry 2022, 13, 828895. [Google Scholar] [CrossRef] [PubMed]
- Kibret, B.G.; Ishiguro, H.; Horiuchi, Y.; Onaivi, E.S. New Insights and Potential Therapeutic Targeting of CB2 Cannabinoid Receptors in CNS Disorders. Int J Mol Sci 2022, 23. [Google Scholar] [CrossRef] [PubMed]
- Legge, S.E.; Jones, H.J.; Kendall, K.M.; Pardiñas, A.F.; Menzies, G.; Bracher-Smith, M.; Escott-Price, V.; Rees, E.; Davis, K.A.S.; Hotopf, M.; et al. Association of Genetic Liability to Psychotic Experiences With Neuropsychotic Disorders and Traits. JAMA Psychiatry 2019, 76, 1256–1265. [Google Scholar] [CrossRef]
- Ishiguro, H.; Horiuchi, Y.; Ishikawa, M.; Koga, M.; Imai, K.; Suzuki, Y.; Morikawa, M.; Inada, T.; Watanabe, Y.; Takahashi, M.; et al. Brain cannabinoid CB2 receptor in schizophrenia. Biol Psychiatry 2010, 67, 974–982. [Google Scholar] [CrossRef]
- Naguib, M.; Xu, J.J.; Diaz, P.; Brown, D.L.; Cogdell, D.; Bie, B.; Hu, J.; Craig, S.; Hittelman, W.N. Prevention of paclitaxel-induced neuropathy through activation of the central cannabinoid type 2 receptor system. Anesth Analg 2012, 114, 1104–1120. [Google Scholar] [CrossRef]
Figure 1.
Sample traces from screen of FDA collection exemplify a PAM hit. Test compounds or vehicle were added at t=1 second and the fluorescence ratio was monitored. Agonist was added at t = 141 seconds. Fluorescence readings for each time point in a well were divided by the fluorescence reading at the initial time point to account for differences in cell number, non-uniform illumination, and dye-loading. The slope value for each kinetic trace was calculated for the time window of 145-155 seconds. The average slope was calculated for wells containing vehicle and this value was subtracted from all wells. Vehicle-subtracted slope was normalized to the relevant maximal agonist signal for each assay. Traces for a sub-maximal agonist response (EC20, green) are compared to a maximal response (ECMax, black) and PAM hit (red).
Figure 1.
Sample traces from screen of FDA collection exemplify a PAM hit. Test compounds or vehicle were added at t=1 second and the fluorescence ratio was monitored. Agonist was added at t = 141 seconds. Fluorescence readings for each time point in a well were divided by the fluorescence reading at the initial time point to account for differences in cell number, non-uniform illumination, and dye-loading. The slope value for each kinetic trace was calculated for the time window of 145-155 seconds. The average slope was calculated for wells containing vehicle and this value was subtracted from all wells. Vehicle-subtracted slope was normalized to the relevant maximal agonist signal for each assay. Traces for a sub-maximal agonist response (EC20, green) are compared to a maximal response (ECMax, black) and PAM hit (red).
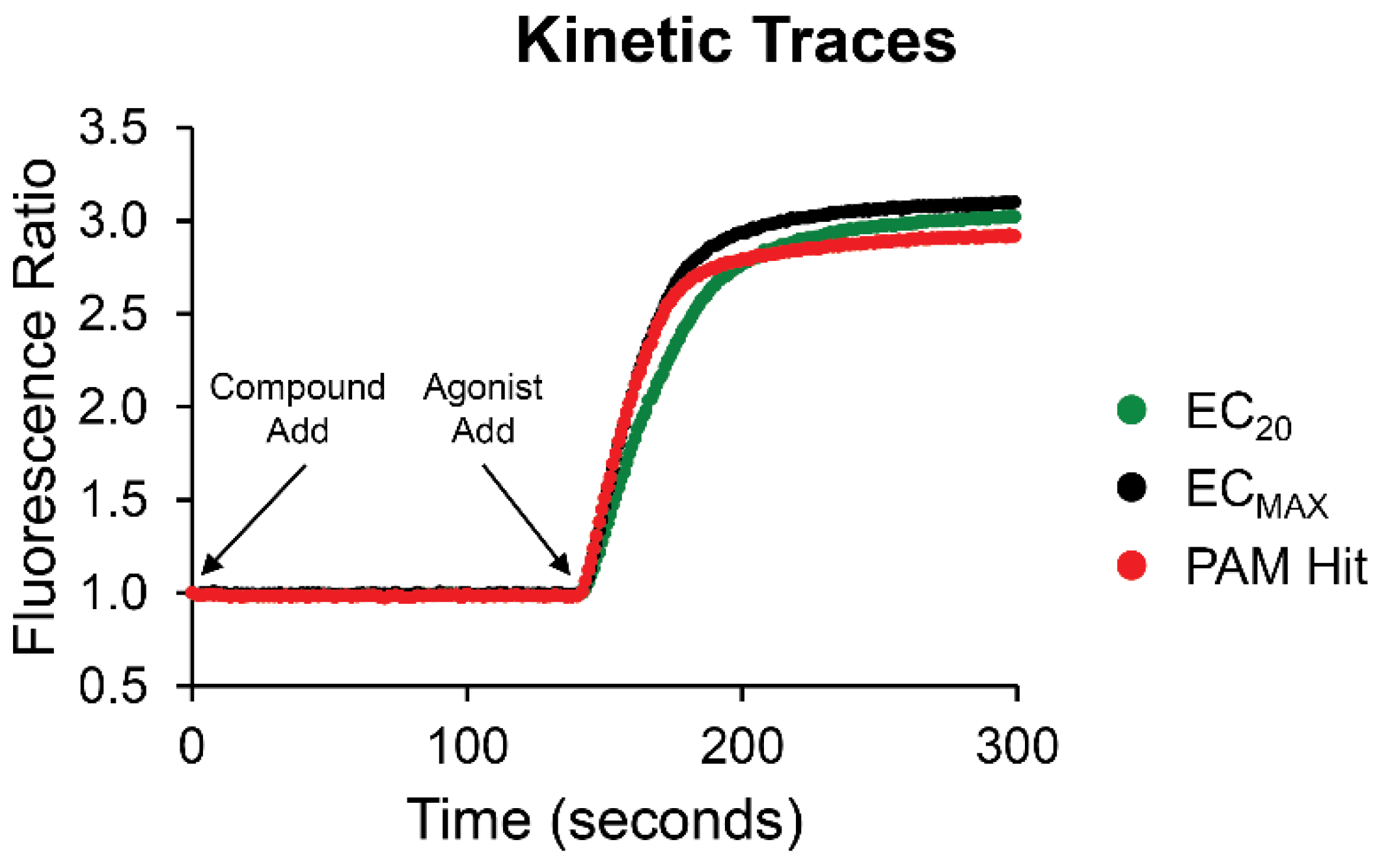
Figure 2.
Mycophenolate mofetil is potent and selective for rCB2 while mycophenolic acid is inactive at CB receptors and parental HEK/GIRK cells. Concentration-response curves in thallium flux assays for (A) mycophenolate mofetil and (B) mycophenolic acid in the presence of a sub-maximal (EC20) concentration of agonist (2-AG or acetylcholine). Results are shown for rCB2 (white), HEK/GIRK (red), and rCB1 (blue) cells. Data represent the mean ± SEM of at least three independent experiments run in duplicate or triplicate, except for mycophenolic acid at HEK/GIRK cells which was run in two independent experiments. Data are plotted as a percentage of maximal 2-AG response for rCB2 and rCB1 or acetylcholine response for HEK/GIRK cells.
Figure 2.
Mycophenolate mofetil is potent and selective for rCB2 while mycophenolic acid is inactive at CB receptors and parental HEK/GIRK cells. Concentration-response curves in thallium flux assays for (A) mycophenolate mofetil and (B) mycophenolic acid in the presence of a sub-maximal (EC20) concentration of agonist (2-AG or acetylcholine). Results are shown for rCB2 (white), HEK/GIRK (red), and rCB1 (blue) cells. Data represent the mean ± SEM of at least three independent experiments run in duplicate or triplicate, except for mycophenolic acid at HEK/GIRK cells which was run in two independent experiments. Data are plotted as a percentage of maximal 2-AG response for rCB2 and rCB1 or acetylcholine response for HEK/GIRK cells.
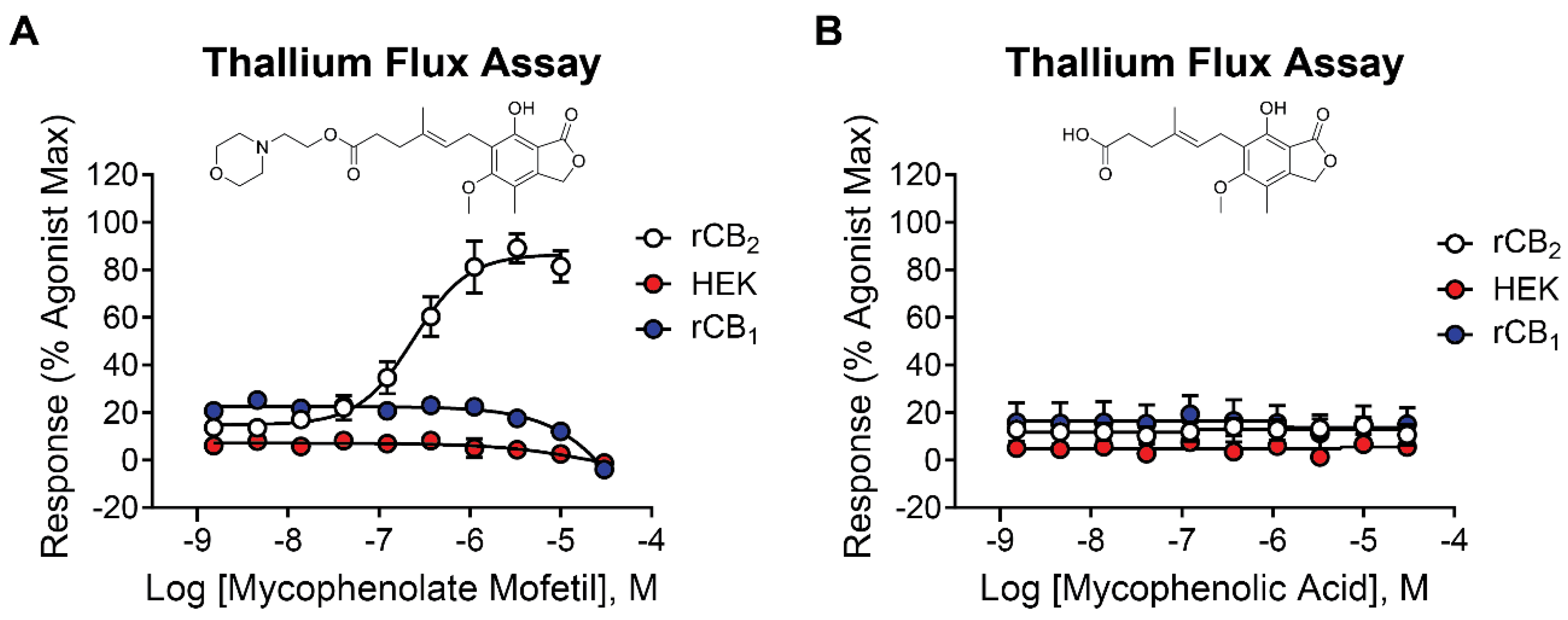
Figure 3.
Mycophenolate mofetil is active at CB receptors in the absence of 2-AG. Concentration-response curves in thallium flux assays for mycophenolate mofetil in the absence of agonist. Results are shown for rCB2 (red) and rCB1 (black) cells. Data represent the mean ± SEM of at least three independent experiments run in duplicate or triplicate. Data are plotted as a percentage of maximal 2-AG response.
Figure 3.
Mycophenolate mofetil is active at CB receptors in the absence of 2-AG. Concentration-response curves in thallium flux assays for mycophenolate mofetil in the absence of agonist. Results are shown for rCB2 (red) and rCB1 (black) cells. Data represent the mean ± SEM of at least three independent experiments run in duplicate or triplicate. Data are plotted as a percentage of maximal 2-AG response.
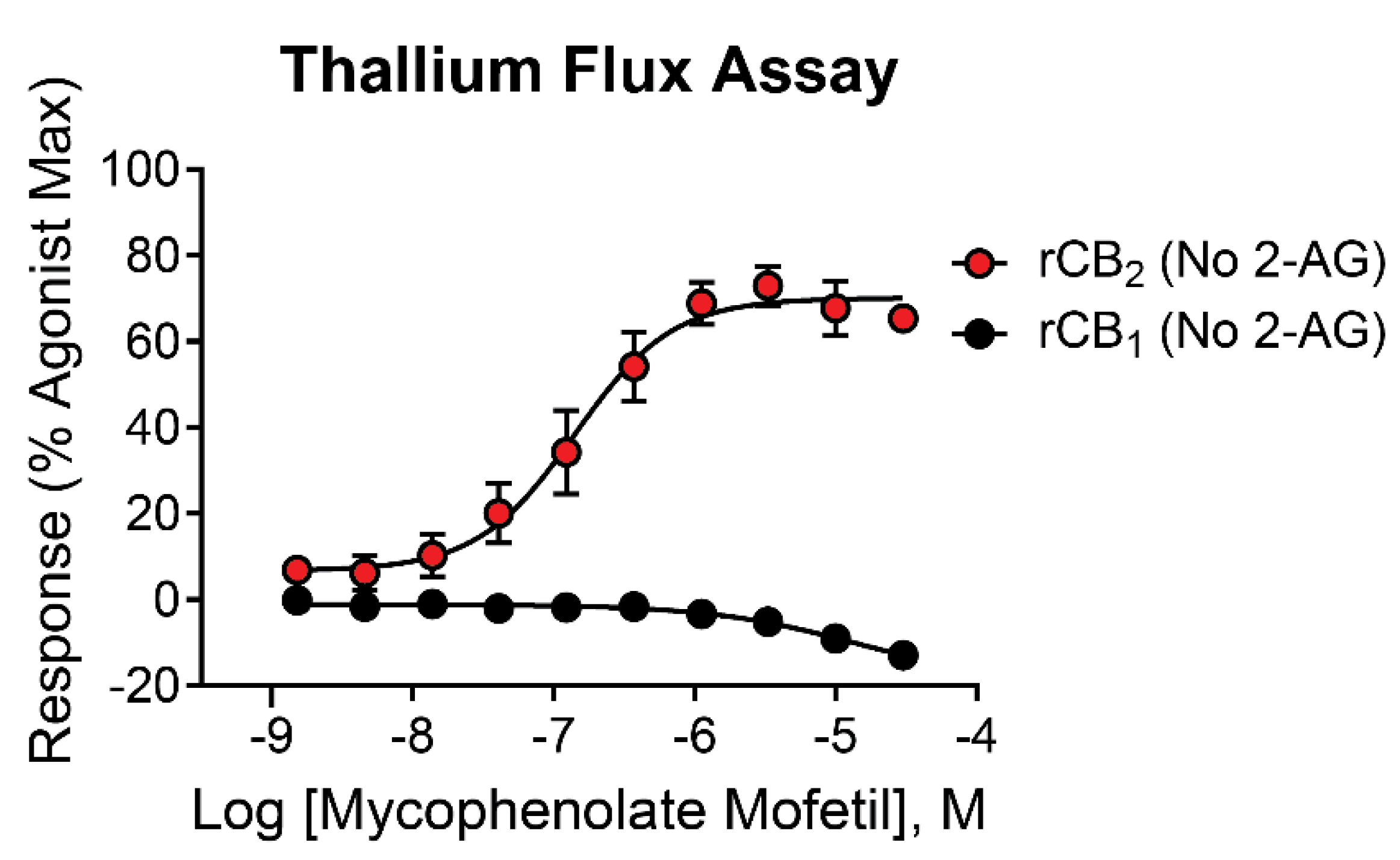
Figure 4.
Increasing concentrations of AM630 shifts the CRC of mycophenolate mofetil in cells expressing rCB2. Concentration-response curves (CRCs) in thallium flux assays for mycophenolate mofetil in the presence of AM630. Data represent the mean ± SEM of at least three independent experiments run in duplicate or triplicate. Data are plotted as a percentage of maximal 2-AG response.
Figure 4.
Increasing concentrations of AM630 shifts the CRC of mycophenolate mofetil in cells expressing rCB2. Concentration-response curves (CRCs) in thallium flux assays for mycophenolate mofetil in the presence of AM630. Data represent the mean ± SEM of at least three independent experiments run in duplicate or triplicate. Data are plotted as a percentage of maximal 2-AG response.
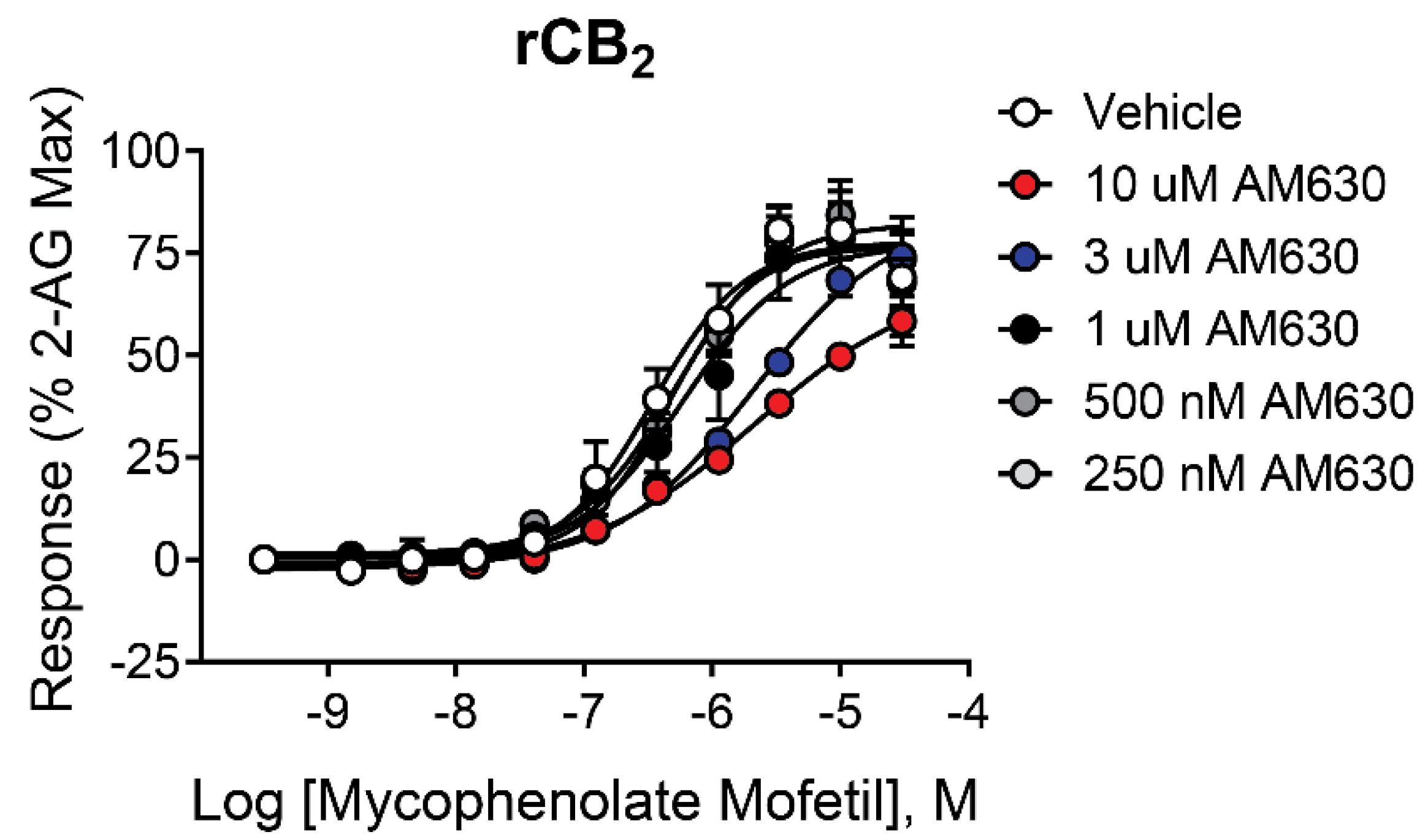
Figure 5.
Mycophenolate mofetil has similar activity in presence and absence of 2-AG in PI hydrolysis. Concentration-response curves in PI hydrolysis assays for mycophenolate mofetil in the presence (white) and absence (red) of a sub-maximal (EC20) concentration of agonist in cells expressing rCB2. Data represent the mean ± SEM of three independent experiments run in duplicate or triplicate. Data are plotted as IP1 concentration in nM.
Figure 5.
Mycophenolate mofetil has similar activity in presence and absence of 2-AG in PI hydrolysis. Concentration-response curves in PI hydrolysis assays for mycophenolate mofetil in the presence (white) and absence (red) of a sub-maximal (EC20) concentration of agonist in cells expressing rCB2. Data represent the mean ± SEM of three independent experiments run in duplicate or triplicate. Data are plotted as IP1 concentration in nM.
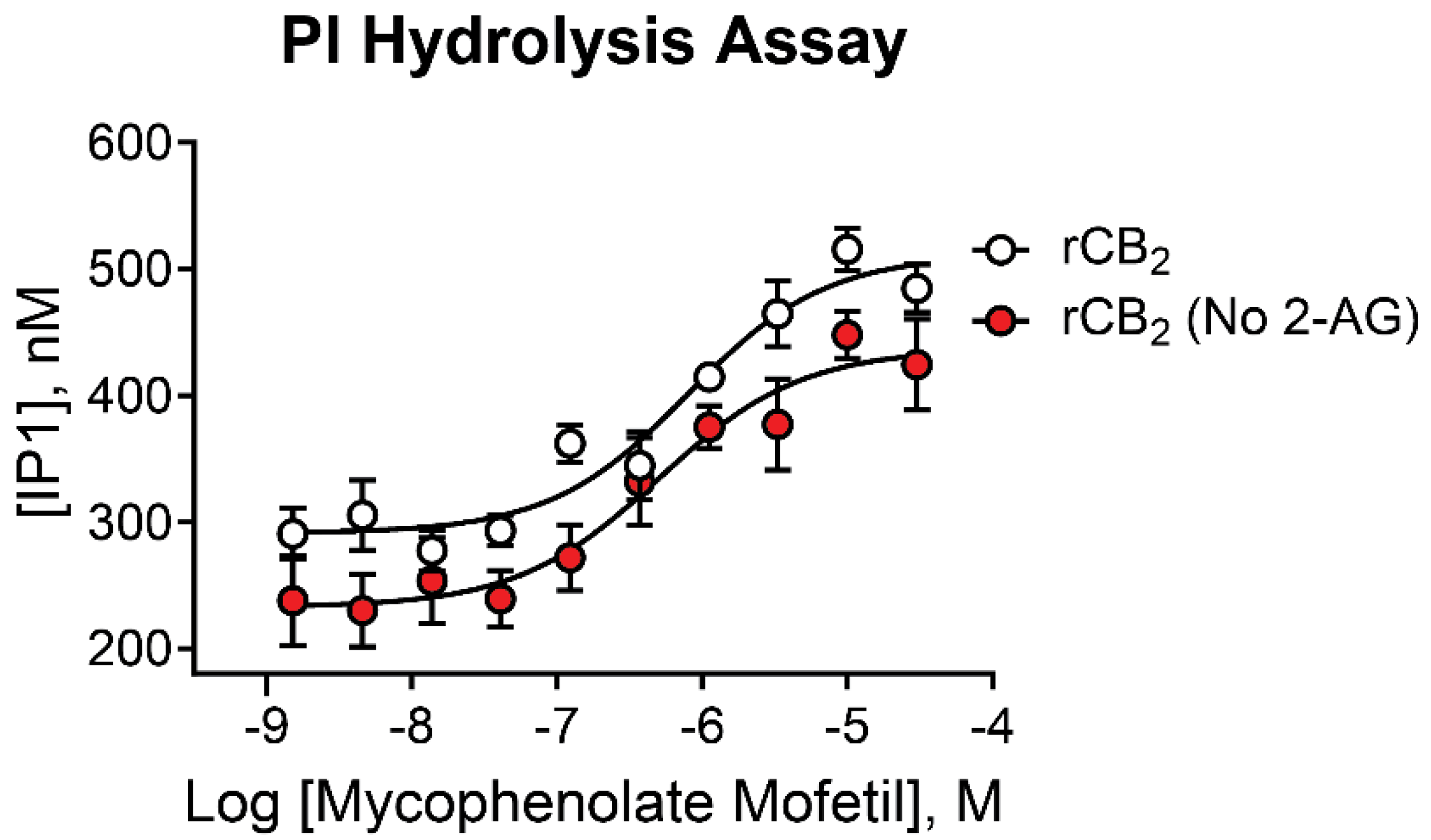
Figure 6.
The CB2 agonists GW-842,166X and MDA7 exhibit distinctions in pharmacological activity in rCB2 thallium flux assays. Concentration-response curves in thallium flux assays are shown for GW-842,166X (A) and MDA7 (B). Results are presented for rCB2 in the presence (white) or absence (red) of a sub-maximal (EC20) concentration of agonist. Results are presented for rCB1 in the presence (blue) or absence (black) of a sub-maximal (EC20) concentration of agonist. Data represent the mean ± SEM of three independent experiments run in duplicate or triplicate. Data are plotted as a percentage of maximal 2-AG response.
Figure 6.
The CB2 agonists GW-842,166X and MDA7 exhibit distinctions in pharmacological activity in rCB2 thallium flux assays. Concentration-response curves in thallium flux assays are shown for GW-842,166X (A) and MDA7 (B). Results are presented for rCB2 in the presence (white) or absence (red) of a sub-maximal (EC20) concentration of agonist. Results are presented for rCB1 in the presence (blue) or absence (black) of a sub-maximal (EC20) concentration of agonist. Data represent the mean ± SEM of three independent experiments run in duplicate or triplicate. Data are plotted as a percentage of maximal 2-AG response.
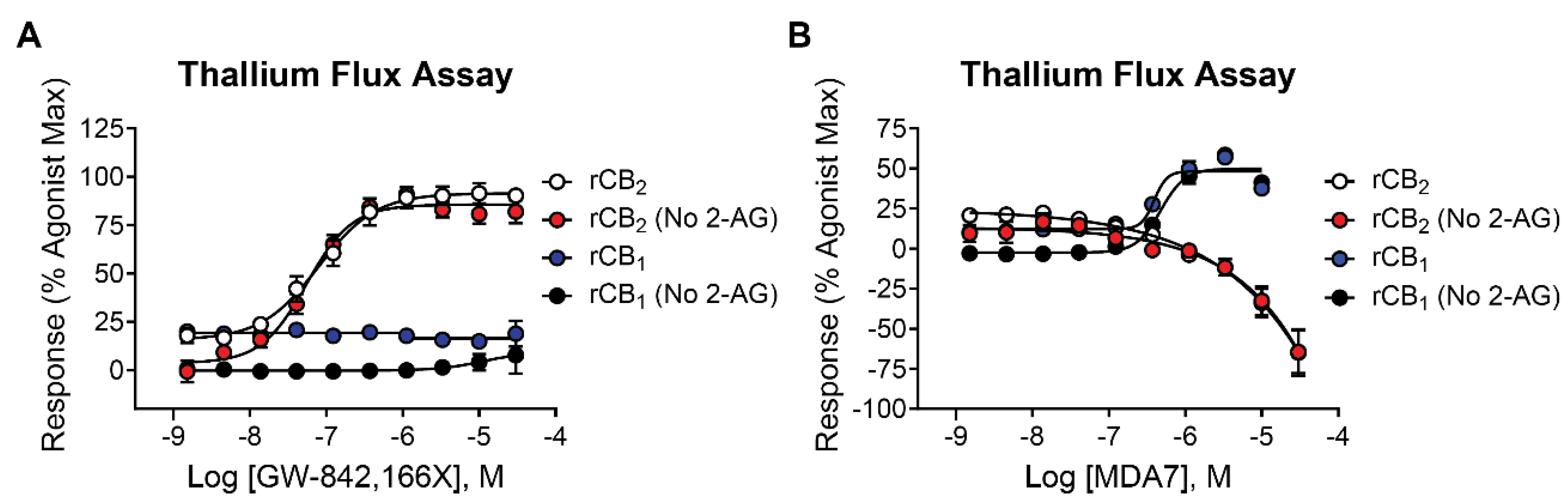
Figure 7.
CB receptor ligands exhibit varying effects on [3H]CP55,940 binding under equilibrium conditions. Competition binding concentration-response curves were obtained in the presence of 0.5 nM [3H]CP55,940 using membranes harvested from HEK/GIRK cells expressing rCB2. Data represent the mean ± SEM of three independent experiments run in triplicate. Data are plotted as a percentage of specific [3H]CP55,940 binding. Non-specific binding was determined in the presence of 10 µM CP55,940.
Figure 7.
CB receptor ligands exhibit varying effects on [3H]CP55,940 binding under equilibrium conditions. Competition binding concentration-response curves were obtained in the presence of 0.5 nM [3H]CP55,940 using membranes harvested from HEK/GIRK cells expressing rCB2. Data represent the mean ± SEM of three independent experiments run in triplicate. Data are plotted as a percentage of specific [3H]CP55,940 binding. Non-specific binding was determined in the presence of 10 µM CP55,940.
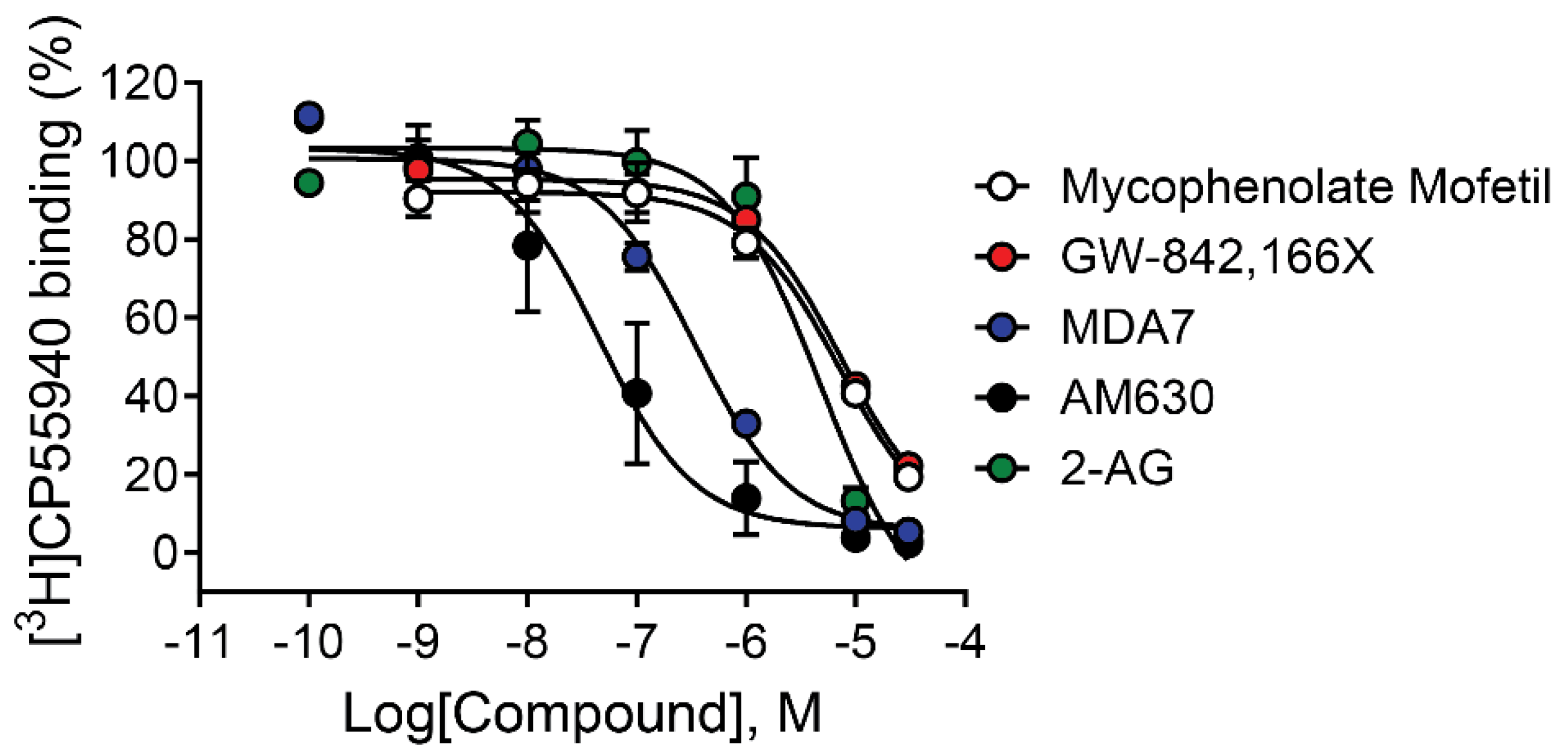
Figure 8.
Differential activity of CB2 agonists in cAMP and β-arrestin assays. Concentration-response curves in cAMP assays in the presence (A) and absence (C) of agonist are shown for CP55,940 (white), mycophenolate mofetil (red), GW-842,166X (blue) and MDA7 (black). Concentration-response curves in β-arrestin recruitment assays in the presence (B) and absence (D) of agonist are shown for CP55,940 (white), mycophenolate mofetil (red), GW-842,166X (blue) and MDA7 (black). Data represent one independent experiment run in duplicate. Data are plotted as a percentage of maximal CP55,940 response.
Figure 8.
Differential activity of CB2 agonists in cAMP and β-arrestin assays. Concentration-response curves in cAMP assays in the presence (A) and absence (C) of agonist are shown for CP55,940 (white), mycophenolate mofetil (red), GW-842,166X (blue) and MDA7 (black). Concentration-response curves in β-arrestin recruitment assays in the presence (B) and absence (D) of agonist are shown for CP55,940 (white), mycophenolate mofetil (red), GW-842,166X (blue) and MDA7 (black). Data represent one independent experiment run in duplicate. Data are plotted as a percentage of maximal CP55,940 response.
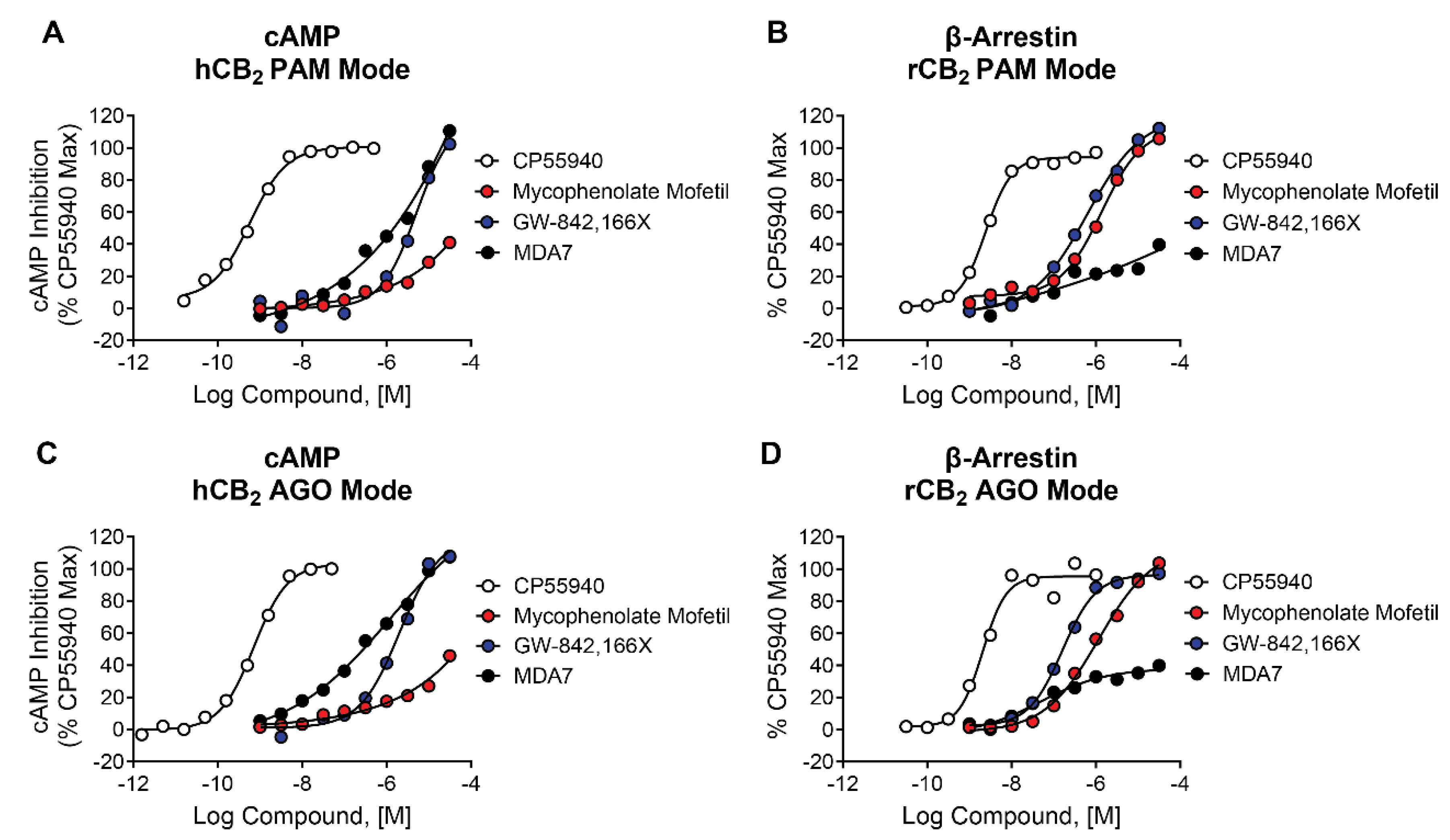

Table 1.
Activity of CB2 compounds in thallium flux and binding assays.
| Compound | rCB2 GIRK | rCB1 GIRK | HEK GIRK | rCB2 Binding | |||
|---|---|---|---|---|---|---|---|
| PAM Activity pEC50 ± SEM % Max ± SEM |
AGO Activity pEC50 ± SEM % Max ± SEM |
PAM Activity pEC50 ± SEM % Max ± SEM |
AGO Activity pEC50 ± SEM % Max ± SEM |
PAM Activity pEC50 ± SEM % Max ± SEM |
Binding Affinity pKi ± SEM % Max ± SEM |
||
mycophenolate mofetil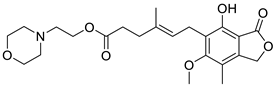
|
6.64 ± 0.06 74 ± 2 % |
6.81 ± 0.12 71 ± 5 % |
< 5.0 -5.5 ± 1.1 % |
< 5.0 -15 ± 3 % |
Inactive | Incomplete pEC50 < 5.0 20 ± 1 |
|
mycophenolic acid 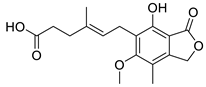
|
Inactive | ND | Inactive | ND | Inactive | ND | |
GW-842,166X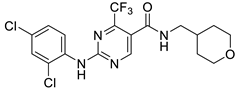
|
7.13 ± 0.08 82 ± 4 % |
7.26 ± 0.05 89 ± 5 % |
Inactive | Inactive | ND | Incomplete pEC50 < 5.0 22 ± 2 |
|
MDA7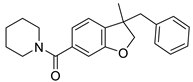
|
< 5.0 -65 ± 15% |
< 5.0 -64 ± 13% |
6.40 ± 0.02 48 ± 2 |
6.32 ± 0.03 50 ± 2 |
ND | 6.60 ± 0.05 5.3 ± 1.7 |
|
*ND indicates not determined.
Table 2.
Activity of CB2 compounds in cAMP and β-arrestin assays.
| Compound | hCB2 cAMP | rCB2 β-Arrestin | ||
|---|---|---|---|---|
| PAM Activity pEC50 % Max |
AGO Activity pEC50 % Max |
PAM Activity pEC50 % Max |
AGO Activity pEC50 % Max |
|
| mycophenolate mofetil | < 5.0 41 |
< 5.0 46 |
5.87 113 |
5.94 114 |
| GW-842,166X | 5.29 119 |
5.69 120 |
6.20 120 |
6.79 97 |
| MDA7 | < 5.0 111 |
< 5.0 108 |
< 5.0 40 |
7.32 38 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



