Submitted:
14 March 2025
Posted:
18 March 2025
You are already at the latest version
Abstract
Despite continuous and extensive global efforts in the fight against tuberculosis (TB) this infectious disease continues to exert a tremendous burden on public health concerns and deaths worldwide. TB, caused by the bacterial species Mycobacterium tuberculosis is highly frequent in people living with HIV. The continuing epidemics with both chronic infections, and the emergence of antimicrobial resistance together with the lack of effective diagnostic tools and drug-drug interactions pose major challenges in the fight against these pathogens. Development of a wide range of host-directed therapies may have the potential to improve treatment outcomes, helping to alleviate the considerable morbidity and mortality associated with both infections. In this review, we discuss the identification and development of new host-directed strategies based on protease inhibitors and their clinical relevance as adjunctive treatment. In the context of therapeutic agents with novel mechanisms, selective protease inhibitors, including saquinavir (SQV) and cystatins (CstC and CstF) are valuable targets that may provide effective therapeutic solutions for control Mtb and HIV coinfection.
Keywords:
protease inhibitors
; saquinavir
; cystatins
; tuberculosis
; HIV-co-infection
; host-directed therapies
1. Introduction
Tuberculosis (TB), primarily caused by the bacterial species Mycobacterium tuberculosis (Mtb), remains a widespread fatal illness and a major public health problem [1]. In 2023, TB has returned as the world’s leading cause of death from a single infectious agent, replacing coronavirus disease (COVID-19), and surpassing the acquired immunodeficiency syndrome (AIDS) deaths caused by the Human immunodeficiency virus (HIV) [1]. During that year, the total number of deaths due to TB was 1.25 million, accounting with 161, 000 demises among people living with HIV [1]. It is estimated that a quarter of the world’s population have been infected with Mtb [2]. However, not all infected individuals will develop TB. With an appropriate immune response some people may completely clear the pathogen or may contain the bacilli in a so-called latent TB infection (LTBI). Confection with HIV is one of the causes that accelerates progression from LTBI to active disease [3].
Since the advent of combined antiretroviral therapy (cART), HIV infection has become a chronic condition. Continuous cART treatment allows the suppression and control of the viral load, thereby preventing the transmission of the infection. According to UNAIDS [4], cART coverage reached approximately 76% of diagnosed individuals with 40 million people living with HIV globally. In 2023, approximately 630,000 people around the world lost their lives to AIDS. It is estimated that at least 1.8 million people have AIDS, which is usually associated with a late diagnosis and treatment of HIV infection. However, recently AIDS has become more prevalent among individuals who have discontinued cART [5]. In addition to the health risks associated with this, the situation increases the risk of HIV transmission and adds to the burden on health systems [6]. TB remains the leading cause of death among people with HIV, responsible for approximately 30% of AIDS-related fatalities.
According to the WHO Global Tuberculosis Report of 2024 [1], Mtb is still responsible for an estimate of a total of 10.8 million new infections. Furthermore, Mtb has been linked with a significant degree of antimicrobial resistance. The proportion of people diagnosed with TB who have rifampicin-resistant TB (RR-TB) and multidrug-resistant TB (MDR-TB, defined as resistance to both rifampicin and isoniazid), collectively referred to as MDR/RR-TB accounts for approximately 5-10% of the total TB caseload (400, 000 cases globally) [1].
A major challenge in the pursuit of an HIV cure is the emergence of drug resistance during cART, primarily attributable to the high mutation rate of HIV, the prolonged duration of treatment, and inadequate adherence to therapy [7]. The emergence of drug-resistant HIV variants has the consequence of compromising the effective inhibition of viral replication by antiretroviral drugs.
In this review, we discuss the identification and development of new host-directed strategies based on protease inhibitors and their clinical relevance as adjunctive treatment for the control of both chronic infections particularly in the context of a coinfection
2. Immuno-Pathogenesis of Mtb-HIV Coinfection
Mtb is a facultative intracellular pathogen which can reside and replicate within macrophages [8]. These cells are considered host professional phagocytes designed to destroy microorganisms within lysosomes. However, Mtb is able to subvert this pathway and survive in immature endosomal vesicles [9]. At the early stages of infection, the surface signatures of Mtb, known as pathogen-associated molecular pattern (PAMPs), are recognized by members of innate pattern recognition receptors (PRRs), particularly from immune cells such as alveolar macrophages and dendritic cells [10]. Upon interaction with PAMPS, there is activation of intracellular signaling pathways which induces the production of pro-inflammatory cytokines, such as tumor necrosis factor α (TNF- α), interleukin-1 (IL-1), IL-6, IL-12, and chemokines all relevant in orchestrating the appropriate defence mechanisms [10,11].
The process of Mtb transmission is initiated by the inhalation of respiratory droplets, which subsequently trigger the activation of alveolar macrophages (AM). These macrophages function as the primary line of defence, engulfing the bacteria and thereby controlling the infection. Furthermore, they act to prevent the dissemination of bacteria to other anatomic compartments [12]. Following this initial phase, pro-inflammatory cytokines are released by these AM as well as by recently arrived macrophages. These cytokines then initiate the formation of innate granulomas, which represent the earliest mechanism of defence against Mtb [13,14] (Figure 1).
Following the innate phase, the adaptive immune responses are founded in CD4+ and CD8+ T-lymphocytes that reacts against Mtb infected cells within the granuloma structure [15]. These cells are important for the activation of macrophages, to a more bactericidal M1 inflammatory state and/or by exerting cytotoxic effects on Mtb infected cells [10] (Figure 1). Altogether this leads to the formation of an adaptive granuloma, a structure that is much more effective in containing bacteria in the lungs than the early innate granuloma [15,16]. A caseating granuloma, in which epithelioid macrophages surround a necrotic core of foamy infected macrophages, is a defining characteristic of TB [15,17]. TB granuloma is inhabited by a plethora of cell types in addition to macrophages, such as neutrophils, dendritic cells (DC), B and T cells, fibroblasts, natural killer (NK) cells and cells secreting extracellular matrix components [18]. Uninterrupted secretion of chemokines by activated infected macrophages attracts neutrophils, monocytes, and lymphocytes, thereby feeding the granuloma with a continuous new arrival of immune cells.
HIV is a master at suppressing the immune response of the host with the potential to be the major cause of Mtb growth and reactivation of LTBI to TB [19]. Individuals infected with HIV are more susceptible to infection with Mtb, partly due to CD4+ T-cell depletion by apoptosis induced by the viral infection that alters their effective immune response [20]. In the case of immunocompetent individuals, a concerted response from several immune cells is observed, working together to target Mtb infected cells and, as previously referred, CD4+ T lymphocytes play a significant role in the control of the infection. CD4+ T-cells function as both helper cells, producing interferon γ (IFN- γ) or IL-17 required to control Mtb infection, and as drivers of the formation of cytotoxic cells (CTLs) from CD8+ lymphocytes, as well in B-cells’ cooperation in the production of antibodies [20]. Consequently, the depletion of CD4+ T-cells influences the number of activated macrophages, leading to the persistence and multiplication of Mtb in the body. The overall reduction of CD4+ T-lymphocytes in the body, together with the entrance of HIV-infected CD4+ T-lymphocytes in granulomas that causes the apoptosis of newly infected cells, is a major cause for the disorganization of the granuloma [21]. This impairment of the granuloma as a solid structure allows for the easy spread of Mtb within the body, thus weakening the body’s control over LTBI and facilitating the progression to active TB infection (Figure 1).
The coinfection of Mtb with HIV therefore appears to be the most significant risk factor for the progression to active TB. As mentioned previously, there is antibiotic therapy for treating TB and an established antiretroviral therapy (ART) to control chronic HIV infection, but the growing resistance to both treatments as well as interactions between the various classes of drugs used are raising serious concerns for effective control of these infections [15,20,22,23,24,25,26,27]. Therefore, there is an imperative requirement for a more profound and detailed understanding of the pathways underlying Mtb-HIV interactions, with an urgent need to develop comprehensive strategies and new efficacious therapeutics to address the challenges posed by TB and TB-HIV coinfection.
3. Current Therapeutic Approaches in TB and During HIV Coinfection
The first-line therapeutics for pulmonary TB, which is the most common form of TB, are Rifampin, Isoniazid, Pyrazinamide, and Ethambutol (RIPE). This RIPE therapy is administered over a period of 6 to 9 months, starting with an intensive phase of 2 months, followed by a continuation phase of 4 or 7 months [28]. This well-planned approach tackles TB, reducing the chances of relapse and drug resistance. However, in cases involving drug-resistant strains of Mtb, the employment of second-line agents, such as injectable aminoglycosides, injectable polypeptides, oral and injectable fluoroquinolones and Bedaquiline is imperative [29].
Rifampin can be administered either orally or intravenously. It is able to inhibit microbially encoded DNA-dependent RNA polymerase via interaction with the rpoB-encoded β subunit. This interaction prevents RNA synthesis by blocking the elongation of the RNA transcript once it extends two to three nucleotides in length [30,31]. Consequently, bacterial strains that possess chromosomal mutations in the rpoB gene, which encodes the rpoB-encoded β subunit of DNA dependent RNA polymerase exhibit resistance to Rifampin treatment [31].
Another drug, Isoniazid, is a prodrug that is activated within the cell following incorporation into the cell and catalysis by the enzyme KatG. This results in the generation of radicals that disrupt the production of mycolic acid, an essential component of the Mtb cell wall [32]. In addition, Isoniazid is activated by NADH and InhA, producing Isoniazid-NADH adducts that inhibit the InhA, a key enzyme involved in mycolic acids synthesis [33]. Selective point mutations in KatG, resulting in diminished ability to activate this drug in bacterial strains, lead to the development of resistance to Isoniazid [32]. A similar mechanism occurs with Pyrazinamide, a prodrug that is activated to pyrazinoic acid by the action of the pyrazinamidase enzyme, encoded by the pncA gene in Mtb [34]. Strains of Mtb lacking pyrazinamidase activity therefore develop resistance to Pyrazinamide. In contrast, Ethambutol exerts its effect by inhibiting arabinosyl transferase, an enzyme which is encoded by the bacterial gene embCAB [35]. Mutations in the embB gene have been associated with resistance to Ethambutol, and it has also been demonstrated that Ethambutol, when paired with beta-lactams, enhances their efficacy against Mtb by improving beta-lactams access through the cell wall [36].
Strains of Mtb that are resistant to both Isoniazid and Rifampin (multidrug-resistant tuberculosis -MDR-TB), treatment with second-line drugs is necessary [37]. Extensively drug-resistant tuberculosis (XDR-TB) strains are characterized by resistance to Isoniazid, Rifampin, a fluoroquinolone, and at least one of the following second-line agents: amikacin, capreomycin, or kanamycin, or otherwise bedaquiline or linezolid [29].
In the context of immunocompromised individuals, such as those infected with HIV, the emergence of drug-resistant strains of Mtb has become a particularly pertinent concern. The management of TB becomes significantly more complex in these cases due to the heightened risk of developing TB in HIV-positive individuals. This imposes regular testing for both LTBI and active TB infection. Another possible complication may include the occurrence of immune reconstitution inflammatory syndrome (IRIS) in individuals undergoing highly active antiretroviral therapy. This condition involves a dysregulated, hyper-inflammatory response against opportunistic infections, usually occurring in the first 6 months of treatment of HIV infected individuals [38,39,40]. The coinfection of Mtb and HIV can be managed through a multifaceted approach, including the combination of ARTs and anti-mycobacterial antibiotics, as well as the prevention of IRIS [41]. However, it is crucial to acknowledge the sophistication of this treatment protocol and the potential for drug-drug interactions (DDIs) between the different therapeutic agents. Moreover, the duration of treatment should be taken into account, preferentially adopting shorter regimens. In addition, TB prophylactic treatment for individuals with HIV should be tailored according to their ART regimen, in order to enhance effectiveness and to limit the adverse effects related to the coinfection [42].
The Center of Disease Control (CDC) currently recommended a 12-week once weekly regimen of Isoniazid and Rifapentine with ART in the absence of any DDIs [42]. Nonetheless, it is imperative to take special consideration when prescribing this combination due to its potential DDIs. This includes drugs like Rifapentine and Rifampin which are contraindicated in HIV-positive individuals taking all protease inhibitors (Doravirine, Rilpivirine, Bictegravir, Cabotegravir, Elvitegravir, Temsavir, and Lenacapavir) [42,43]. Appropriate ART includes efavirenz once daily or Raltegravir twice daily with either abacavir/lamivudine or tenofovir disoproxil fumarate/emtricitabine. However, Rifampin and Rifapentine significantly reduce the effective doses concentration of these drugs, thus necessitating adjustments to ARTs regimens, which in turn increase the likelihood of drug related adverse effects [42].
The adjustment in dosage of drugs is essential for the maintenance of adequate drug levels, which will provide efficient treatment for both TB and HIV without any risk of failure. For instance, the current guidelines recommend a dose of 600 mg of Rifamycin to be given alone or co-administered with Efavirenz [44], but some sources suggested a higher dose of 800 mg for individuals with a body weight of more than 60 kg [44]. It is important to note that dolutegravir is not recommended in patients with integrase strand transfer inhibitor resistance, though it can also be used as a first line ART [42]. Rifabutin is another drug in the Rifamycin family which has shown its efficacy during Mtb treatment [45]. An alternative option is to consider shorter drug courses, which are gaining popularity over traditional regimens with higher efficiency and greater completion [46]. The BRIEF TB/AF279 clinical trial demonstrated the efficacy of an ultra-short regimen comprising a 1-month course of Rifapentine and Isoniazid, achieving a higher completion rate than the 9-month course of Isoniazid in preventing TB infection in HIV-infected individuals [47]. This regimen is now recommended by the WHO [47,48]. In addition, the development of several compounds has also been reported, including a family of N-alkyl nitrobenzamides that demonstrate promising antitubercular activities and even can be classified as a structural simplification of previously known inhibitors of decaprenylphosphoryl- β-D-ribofuranose 2′-oxidase (DprE1), a critical enzyme of Mtb and a new antitubercular target [49].
In HIV-positive patients with active TB infection, CD4+ T-cell counts are a crucial factor in determining the initiation of ART [42]. On the basis of the CD4+ T-cell counts, the starting of ART should be initiated within two weeks of TB treatment initiation if the level is less than 50 cells/mm3, while it can be commenced within eight weeks of TB treatment initiation if the CD4+ T-cell count is 50 cells/mm3 or above [42,43]. Two main treatment options are currently available for individuals with active TB and HIV infection. The first option involves a six-to-nine-month period of intensive treatment with the combination drug, Rifampin, Isoniazid, Pyrazinamide, and Ethambutol for two months followed by a four-month continuation phase of rifampin and isoniazid [50]. The second option consists of a 4-month regimen of Rifapentine-Moxifloxacin, which can cater to patients whose CD4+ T-cell count is 100 cells/microliter, and an ART regimen such as Efavirenz [51]. Additionally, a number of novel therapeutic approaches for countering the co-infection have been proposed, including the use of dual targeted anti-HIV/anti-TB heterodimers [52]; and the targeting of HIV’s inhibition of NK cell-mediated immunity by HIV in response to Mtb infection [53].
Despite the advancement and development of therapeutic approaches to treat and control TB and chronic HIV infection, the synergistically action of these pathogens, rising resistance to both treatments, and drug-drug interaction, are leading to serious diagnostic and therapeutic challenges. Coinfection with Mtb increases the risk of death of HIV-positive patients, while late-stage HIV infection increases the risk of developing TB in latently infected individuals. Nevertheless, there remains an ongoing and critical demand for complementary management approaches that can bypass the constraints of the existing antimicrobial treatment portfolio. A great variety of host-directed therapies (HDTs) with various mechanisms of action could be identified and developed to serve as adjuncts to standard treatment for infections caused by bacteria and virus.
4. Host-Directed Therapies: Current Status and Recent Progress for the Treatment of Infections
From an evolutionary perspective, host-pathogen interactions can be understood as states in which microbes exist without causing harm or manifesting overt disease [54,55]. In fact, the action of the host’s innate and adaptive immune surveillance mechanisms determines whether the infection will be contained or whether it will progress to clinical disease resulting in recovery or death [54]. It is critical to acknowledge the pivotal role that host-related events play in the outcome of antimicrobial treatment, particularly in light of the fact that disease progression after infection can result in tissue damage, leading to long term functional impairment and increased mortality. Among these are immune dysregulation from various origins, including, but not limited to, an excessive inflammatory response to infection, the use of immunosuppressive medications, disadvantageous socioeconomic conditions, HIV infection or the presence of comorbidity including non-communicable diseases such as diabetes, neoplasia, and chronic pulmonary disease [56].
In the context of infectious diseases, it is crucial to understand the complex interplay between the host and the pathogen, in order to elucidate the mechanisms underlying pathogenesis. In the past few years, there has been a shift in scientific research with a transition from a direct targeting of pathogen components to a focus on host factors. This paradigm shift has opened promising new avenues that hold the potential to enhance clinical outcomes, referred to as host-directed therapies. Host-directed therapies (HDTs) are defined as host-directed interventions that can modulate intracellular pathways involved in innate or adaptive immune responses to microbes to enhance immune response and reduce immunopathology [57]. About 90 % of the tested HDTs have demonstrated comparable efficacy levels towards drug resistant and drug susceptible pathogens [58]. In addition to their efficacy in battling drug resistant pathogen, HDTs have been hypothesized to reduce the likelihood of developing drug resistance, by targeting multiple cellular and intracellular mechanisms that are critical for microbial replication and pathogenesis [58].
HDTs can also enhance the efficacy of anti-microbial treatments. A recent study evaluated 183 host-directed drugs, of which 30% were found to have activity against more than one pathogen. The majority of these (52%) targeted close evolutionary relatives, while 48% were active against evolutionarily distinct groups at the levels of families, kingdoms, and domains [58,59]. The HDTs comprise drugs with different mechanisms of action. These include agents that enhance host immunity, such as CAR-T cells, which have been used in the treatment of HIV-1-infected individuals, resulting in a transient decrease in the HIV-1 viral reservoir [60]. Administration of type I interferon (IFN) has also been used for the treatment of chronic Hepatitis C (HCV) infection or COVID-19 [61,62,63]; N-acetylcysteine, an antioxidant to protect cells from oxidative damage in TB [64]; anti-pathogen antibodies used for COVID-19 [65] and respiratory syncytial virus infection (RSV) infection [66].
HDTs that are predicted to diminish immunopathology by attenuating inflammation caused by the immune response against the pathogen comprise the compounds targeting cytokines, such as Interleukin (IL)-6 receptor blockade in COVID-19 [67], tumor necrosis factor (TNF) treatment in tuberculosis meningitis [68] and Janus kinase (JAK) inhibitors in COVID-19 [67]. Other potential HDTs include the use of corticosteroids in COVID-19 and tuberculosis meningitis [67,69], anti-oxidants such as N-acetylcysteine in tuberculosis [64]; Vitamin D to decrease inflammation in TB of the lung, and certain anti-inflammation drugs (statins and cyclooxygenase 2 inhibitor in TB) [70,71,72].
HDTs are also expected to modulate the immune response to Mtb, thus emerging as a compelling adjuvant therapeutic strategy. There is mounting evidence to suggest that several facets of the immune response including reactive oxygen species production, antimicrobial peptide synthesis, cytokines production, autophagy induction, and cell-mediated immunity can be modulated by HDT to hinder Mtb growth (Figure 2A). One of the potential host therapeutic targets against Mtb includes the impairment of granuloma integration through neutralization of TNF-α by using Enbrel (anti- TNF-α antibody). This disruption enhances the bacterial elimination via antibiotic therapy and curtails lung pathology [73](Figure 1). Another recent clinical trial has demonstrated that etanercept-mediated TNF blockade increases CD4 cells amongst patients with HIV-associated TB in the early treatment phase of TB. Nevertheless, anti- TNF-α antibody therapy can also exacerbate the severity of the disease by supressing the immune response of the host, as has been established in animal models [73,74].
Moreover, HDTs can modulate immune response by targeting the vitamin D signalling pathway, which is pivotal in augmenting Mtb-killing activity of macrophages. Vitamin D has been shown to induce autophagy through the down-regulation of the mTOR protein [75]. Additionally, CAMP gene expression can be activated by the co-administration of phenylbutyrate with vitamin D3 and standard anti-TB drug therapy and has been shown to inhibit the intracellular growth of Mtb [76,77]. A recent study found that vitamin D at 1.25 mg supplementation with standard first-line anti-TB drugs corrected the vitamin D deficiency, but no significant change in the Mtb culture conversion in pulmonary TB has been observed [78,79]. A similar finding was reported for sputum conversion, which was improved with vitamin D supplementation in TB patients with vitamin D receptor “tt” genotype [79]. These conflicting results may be attributable to differences in dose, duration of treatment, stage of disease and other factors. Therefore, further adequately sized studies in appropriate clinical conditions are required to understand the epidemiological effects of vitamin D supplementation in TB.
HDTs as a potential adjuvant in the treatment of TB also include drugs that targets the anti-inflammatory response, such as aspirin and ibuprofen [80] (Figure 2A). Aspirin appears to exert a greater control over bacillary load, translating into better survival in advanced disease as shown in a study conducted in the C3HeB/Fej murine model [81]. In a different study, the administration of, aspirin to BALB/c mice potentiated the action of pyrazinamide, leading to enhanced clearance of viable mycobacteria in both the spleen & lungs. However, the combination of aspirin and Isoniazid resulted in increased bacterial loads [82]. Collectively, these findings indicate that further clinical studies should be conducted to evaluate the therapeutic effect of concomitant aspirin administration when combined with other anti-TB drug combinations. In a mouse model mimicking active TB, the bacillary load was also decreased by ibuprofen treatment [83]. Additionally, in a different study performed in a murine model, ibuprofen proved to potentiate the effect of pyrazinamide in TB treatment [82].
It is anticipated that HDTs should improve cell-mediated immunity. Such agents include cholesterol-reducing drugs such as statins. Several mechanisms against TB have been identified, which include the inhibition of foamy cell formation, supporting Mtb persistence via reversing cholesterol biosynthesis [84] and the induction of phagosomal maturation and autophagy [85]. For instance, the autophagy inhibitor 3-methyladenine has been shown to reverse autophagy and the maturation of phagosomes. Conversely, the administration of the statin simvastatin at a dose of 25 mg/kg has been shown to accelerate the clearance of bacteria in the lungs of mice when used in conjunction with standard therapy [86,87]. However, a large retrospective study utilizing a national medical claims database revealed that statins did not offer protection against tuberculosis in patients with diabetes mellitus [87,88], highlighting the need for further research to determine the most effective agent and dosing schedule for clinical trials. Other mechanisms involve an increase in the percentage of Natural killer T (NKT) cells in the cultures and up regulation of co-stimulatory molecules on monocytes, along with higher IL-1β and IL-12p70 secretion [89,90] and inhibition of TGF- β [91,92].
A variety of evidence and studies, as well as the number of on-going interventional clinical trials, have nicely revealed HDTs were nicely revealed as a promising solution by repurposing known effective drugs and targeting new candidates. Future interventions will be based on new strategies using novel host-directed strategies identified and developed through protease inhibition (Figure 2B).
5. Protease Inhibitors as a Strategy to Control Infectious Diseases
Enzymes have long been recognized as ideal candidates for target-based drug development, as evidenced by decades of research on disease agent proteases. Targeting selective protease inhibitors has been demonstrated to deliver effective therapeutic solutions for the treatment of some of the most important infectious diseases like malaria and COVID-19 [93]. Validated proteolytic targets for the infectious diseases include proteasomes of the malarial pathogen Plasmodium falciparum, P. falciparum aspartyl proteases and SARS-CoV-2 viral proteases [93]. Furthermore, low-molecular-weight inhibitors of proteases recently have emerged as promising therapeutic agents in the treatment of numerous diseases, including hypertension, diabetes, and various cancers [94]. Indeed, proteases play crucial roles in nearly all biological processes, both within individual organisms and extracellular events. This generally involves either the activation of other enzymes through their well-timed processing, resulting in the generation of active, accessible catalytic sites on substrates, or conversely the inactivation of proteins through their proteolytic fragmentation [95]. There are seven classes of proteases, which mainly include metallo-, serine, aspartyl, cysteine and threonine proteases [96]. Among them, serine proteolytic enzymes are found in the highest natural abundance is for serine proteolytic enzymes, followed in decreasing order, by metallo-, cysteine-, aspartate and threonine proteases [97].
Proteolytic enzymes are considered to be a primary component of the major virulent factors of infectious agents, playing a pivotal role in their development, reproduction and interactions with host/invertebrate vector tissues. Therefore, these enzymes are regarded as promising targets for designing new drug candidates for the treatment of infectious diseases [98,99]. In (1995), the discovery of HIV protease inhibitors (HIV-PIs), such as Saquinavir, Lopinavir and Ritonavir [100,101], demonstrated the antiviral efficacy of enzyme-targeted drugs. These HIV-PIs inhibit viral replication by selectively blocking the retroviral protease, thereby preventing the processing of the long polypeptide that the viral RNA genome encodes into individual viral proteins [102]. The advent of novel inhibitors and their combinations have been fundamental for the development of effective and less toxic antiretroviral therapy (ART), transforming HIV infection from a fatal to a more manageable disease. The development of protease inhibition-based therapy has emerged as an attractive and potential strategy against infections since the discovery and approval of HIV-PIs. For instance, the papain-like cysteine protease cruzain has been demonstrated to be essential for the life cycle and virulence of Trypanosoma cruzi, the causative agent of Chagas disease [103]. Likewise, an irreversible cruzain inhibitor, vinyl-sulfone, has shown efficacy against schistosomiasis, hookworm infections and cryptosporidiosis [103]. As a therapeutic strategy for multiple myeloma treatment, selective proteasome inhibitors such as carfilzomib, bortezomib and ixazomib have been developed [104]. In addition, the development of specific inhibitors is critical in the treatment of infectious diseases such as malaria, leishmaniasis, schistosomiasis and Chagas disease [105].
Another example validating protease-inhibition-based drug development can be seen in the repurposing of drugs designed to target COVID-19. These candidate compounds inhibit the SARS-CoV-2 main protease (Mpro), which is known to play a crucial role in the virus replication process in infected cells [106]. The PLpro protease of SARS-CoV-2 shares high sequence and structural similarity with PLpro of previously emerged coronavirus (SARS-CoV-1) and has been characterized as displaying the same functions in virus replication and modulation of the host’s immune responses [107]. Thus, the inhibition of SARS-CoV-2 protease can block viral replication and enhance the innate immune responses in acute COVID-19 infection. Several studies have demonstrated repurposing of SARS-CoV-1-PLpro drugs against SARS-CoV-2-PLpro [107,108,109].
As previously mentioned, saquinavir (SQV) controls HIV infection by interfering with HIV protease activity, ultimately preventing virion infectivity and intercellular transfer. It was hypothesized that SQV could be repurposed to regulate a variety of protease-dependent infectious organisms. The proteostatis network, comprising chaperones, proteases and proteasome, plays a major role in the survival of Mtb under cognitive host immune stress [110]. HIV and Mtb interact synergistically in their co-infected hosts, modifying the host immune environment, thereby promoting the replication and dissemination of both pathogens. Hence it is plausible that during Mtb infection, particularly in the context of Mtb/HIV coinfection, HIV PI could interfere with the host proteases. The potential and proven proteolytic targets against a broad range of infectious diseases provide compelling evidence for the establishment of protease inhibitors as a drug development platform and other therapeutics strategies. Overall, a more profound understanding of microbial dependence on cellular proteases and their inhibitors may establish a strong basis for designing novel host-directed strategies to control microbial infection.
6. New Host-Directed Strategies Based on Proteases Inhibitors for Mtb and Coinfection with HIV
This section will focus on protease enzymes, which have been putatively associated with proteolytic activity in Mtb-infected macrophages as well as in case of coinfection with HIV. Previous studies have already demonstrated that Mtb induces a general down-regulation of lysosomal proteases, which ultimately helps Mtb to establish its intracellular niche within macrophages [111,112]. In the context of coinfection with HIV, the synergistic effect between HIV and Mtb, leads to the emergence of this niches as important mediators of the infection’s pathogenicity. This may represent a mechanism by which pathogens can manipulate the host’s microbicidal responses, thereby facilitating their intracellular survival within the immune cells. In this scenario, it is possible that HIV PIs, including SQV, could be repurposed to control infections caused by these proteases-dependent pathogens. This hypothesis is further substantiated by the fact that SQV and other PIs have already shown inhibitory effects against various pathogens, including Plasmodium falicparium [113], Trypanosoma cruzei [114] or SARS-CoV [115].
6.1. Repurposing Saquinavir as a Host-Directed Strategy to Control Mycobacterium Tuberculosis Infection
A plethora of antiretroviral drugs are already available for the treatment of HIV infection, but only a small number of these (PIs) have the capacity to interfere with virus production and release from macrophages during chronic infection. It has been demonstrated that HIV PIs, particularly SQV, may interfere with the proteases of the host during Mtb or Mtb/HIV infection. In this context, SQV,a first-generation HIV PI, SQV, has been repurposed as a host-directed therapy for tuberculosis, particularly in the context of coinfection with HIV [116]. In this study, it has been demonstrated that SQV rather than acting directly on infected macrophages as a PI, can enhance omnicathepsins proteolytic activity during either Mtb mono-infection or during HIV coinfection. Briefly, these endolysosomal enzymes, particularly cathepsin S, are attributed to pathogen destruction as one of the earliest innate immune responses after infection [116]. Cathepsin S is also involved in the antigen presentation machinery and regulation of autophagy [116,117,118,119]. However, Mtb infection has been shown to disrupt these enzymatic activities (cathepsins B, S and L), which are critical for both innate and adaptive immune responses [111,112]. Consequently, the study [116] demonstrated that SQV treatment led to enhanced pathogen killing attributable to heightened cathepsin activity. Additionally, the study validated the role of cathepsin S as part of the antigen- presenting machinery, with marked upregulation of HLA class II molecules on SQV treatment. Similarly, SQV treatment resulted in increased levels of T cell-secreted IFN-γ, which could have activated the bactericidal state in macrophages, leading to improved control of the immunopathology in the lungs [116].
Autophagy is a process that is subject to regulation by cathepsins S and L [120]. It also established that Mtb and HIV have the capacity to inhibit autophagy. Interestingly, upon infection by one pathogen, alveolar macrophages may in fact preferentially sustain the second pathogen or may even induce a similar response in neighbouring cells [121]. Therefore, the SQV-mediated enhancement of proteolytic activities of cathepsins S and L, as confirmed in the study by Pires et al. [116], would also augment autophagy. Consequently, this would facilitate infected cells in the clearance of pathogens and cytosolic aggregates, as well as inflammatory signalling mediators, ultimately resulting in the reduction of tissue inflammation [122]. Furthermore, the study has also demonstrated enhanced internalization of SQV in macrophages using a liposomal drug delivery system, which has a significant impact on the proteolytic activity of cathepsins compared to that of free drug treatment and also without any cytoxicity [123]. In conclusion, there are studies and relevant data that support the conclusion that SQV has a potential as a host-directed therapy for TB.
6.2. Modulation of Cystatins C and F as a Host-Directed Strategy to Control Mtb Mono-Infection or Coinfection with HIV
Several proteolytic enzymes are involved in various physiological processes that are related to the maintenance of homeostasis in host cells [124]. They play important role in the digestion of bacteria that have been taken up via the process of phagocytosis [125]. Mtb are intracellular pathogens with the potential to establish themselves within their primary niche, which is the phagosomes of the host phagocyte, in particular macrophages. To this end, the bacilli hamper phagosomal maturation and downstream intracellular macrophage efficacy [126,127]. This dysregulation of host macrophages is capable of affecting proteolytic enzymes expression like lysosomal cathepsins resulting in an increased intracellular survival of Mtb [111]. In fact, the abnormal activity of proteolytic enzymes needs to be tightly controlled as this could cause severe dysfunction and pathology. Cystatins (Csts) are a well-known class of endogenous modulators of cathepsins, which generally control their excessive enzymatic activity by binding and sequestering them within cells, tissues, and body fluids [124]. It is possible that pathogens, particularly in the case of Mtb and HIV, could evolve strategies for the manipulation of these early events to prevent the activation of the microbicidal mechanisms, allowing the pathogens to survive within the cells. Therefore, Csts may emerge as potential targets that could be manipulated to restore cathepsin activity in the context of Mtb infection or coinfection with HIV.
Csts are well characterized inhibitors which are further divided into four different families. Type I Csts (stefins) consist of CstA and B and exist within the cytosol and nucleus, while type II Csts include CstEM, S, SA, SN, and D, which are secreted and function as extracellular proteins [128,129]. Some of the secreted type II Csts (CstC and F) could be internalized by immune cells, or from the secretory pathway translocated into the cytosol of the immune cells accumulating in endosomal/lysosomal vesicles [130,131]. Type III Csts include kininogens that circulate in the blood as precursors of the vasoactive peptide kinin (Cst families reviewed in [128,129]). Finally, type IV Csts comprise thyropines and are characterized as a Csts family originating from the observation that a fragment of the p41 invariant chain bound to MHC class-II molecules inhibits cathepsin L [132,133].
Cathepsins and their natural inhibitors are regarded as the key players in Mtb and HIV infection. Thus, therapies based on the manipulation of these relevant players could restore the protease-antiprotease balance which in turn can control infection, transmission and excessive inflammatory responses. A transcriptional mapping of the human macrophage transcriptome for type I and type II cystatins during monoinfection or coinfection, revealed CstC as a major target and CstF as a potential target for controlling Mtb or Mtb/HIV infection [134]. The study demonstrated that knocking-down Cst C mRNA significantly improved the intracellular killing of Mtb. In addition, the expression of human leukocyte antigen (HLA) class II in macrophages, the proliferation of CD4+ T-lymphocytes and IFN-γ secretion were increased [134]. This increased IFN-γ production will obviously lead to proinflammatory activation of macrophages with increased microbicidal activities against Mtb [125]. A subsequent study has demonstrated the development of a new potential nanomedicine based on chitosan for delivering a siRNA-targeting cystatin C to the infected macrophages model with significant impact on the intracellular viability of Mtb [135]. Collectively, the results showed that the manipulation of Cst C expression in human macrophage has significant impact on the innate immune regulation of Mtb intracellular growth, as well as on the strengthening of the adaptive immune responses against mycobacterial infection [134].
Furthermore, the depletion of CstF has been demonstrated to modulate the proteolytic activity of the macrophages lysosomal cathepsins (CtsS, CtsL and CtsB) thereby enhancing the intracellular killing of Mtb [136]. Indeed, the depletion of CstF expression has been shown to control Mtb intracellular loads, including multi-drug resistant clinical strains that are resistant to first-line antibiotics used to treat TB (Figure 3).
These findings are consistent with earlier observations using the CstC cathepsin inhibitor, where similar results were obtained in macrophages [134]. Direct regulation for cathepsins L and S was indeed demonstrated via targeting cystatin F expression, as previously reported, while no evidence for direct inhibition of the cathepsin B was found [137,138]. The findings of this study do not distinguish between a direct or indirect role for CstF, which may account for the contradictory results obtained. Overall, these two studies on Cst C and Cst F suggest that the targeting protease inhibitors could be a promising approach to improve the control of Mtb infections, even in clinical strains that are resistant to first-line antibiotics used to treat TB [134,136].
HIV-infected patients frequently exhibit pleural effusion due to TB in the context of coinfection [140]. In addition, higher levels of CstF have been detected in pleural effusion of TB patients compared to other inflammatory conditions [141]. Consequently, CstF could be proposed as a potential target in the context of Mtb/HIV coinfection. It has been hypothesized that the depletion of CstF could augment control over HIV infection, in addition to the already established microbicidal effects over Mtb [139]. Indeed, the analysis revealed that CstF depletion in Mtb-infected macrophages enhances cathepsin C activity in cocultured lymphocytes infected with HIV, which in turn augments their granzyme cytotoxic effects. As a result, a significant impact was observed on HIV replication and viral loads [139](Figure 4). Taken together, the study proposed that CstF may provide a new target for new therapeutic strategies aimed at controlling both Mtb and HIV pathogens.
7. Conclusion and Future Directions
In conclusion, this review highlights the contribution of HDTs to mitigate the high burden of infectious diseases focusing more on mycobacterial infection and Mtb/HIV co-infection. In addition, it addresses novel HDTs based on protease inhibitors and their clinical implications as adjunctive therapeutic regimens to manage TB and HIV coinfection. Among these novel strategies are SQV repurposing (an HIV protease inhibitor) and Csts manipulation (the natural cathepsin inhibitors). The present review emphasizes that HDTs represents an evolving approach to managing bacterial infection, mainly by restoring the compromised host immune responses. The transition from Mtb infection to active disease is influenced by coinfection and immune suppression, as observed in HIV coinfection. Furthermore, HDTs have the capacity to boost immune responses and alleviate immunopathology by targeting host immune and inflammatory pathways, resulting in beneficial effects on outcomes of bacterial and viral treatments. A range of various HDTs that have been developed to combat pathogens by targeting multiple cellular and intracellular mechanisms required for microbial replication and pathogenesis. Consequently, HDTs may also contribute to a reduction in pathogen transmission within the community and improve clinical outcomes. This suggests that HDTs could be a game-changing strategy in the treatment of infectious diseases.
Author Contributions
Conceptualization, M.M. and E.A.; writing—original draft preparation, M.M.; writing—review and editing, M.M., E.A., D.P., and J.M.A.-P.; visualization, M.M. and E.A.; figures: M.M and E.A..; supervision, E.A.. All authors have read and agreed to the published version of the manuscript.
Funding
The research linked to this work was funded by Fundação para a Ciência e a Tecnologia (FCT)—grant numbers PTDC/SAU-INF/28182/2017 to E.A., 2023.14955.PEX (doi:10.54499/2023.14955.PEX) to D.P., and UID 04138 to IMed-ULisboa. It was also funded by Gilead Sciences Portugal (Gilead Génese Programme 2019) to J.M.A.-P.; M.M. is supported by a PhD fellowship from FCT with the reference 2021.07978.BD.
Acknowledgments
The authors thank Associação para o Ensino e Investigação em Microbiologia (ADEIM).
Conflicts of Interest
The authors declare no conflict of interest.
References
- 2024 Global Tuberculosis Report, World Health Organization, 2024. Available online: https://www.who.int/teams/global-tuberculosis-programme/tb-reports/global-tuberculosis-report-2024 (accessed on 14 January 2025).
- Houben, R.M.G.J.; Dodd, P.J. The Global Burden of Latent Tuberculosis Infection: A Re-Estimation Using Mathematical Modelling. PLoS Med 2016, 13, e1002152. [Google Scholar] [CrossRef] [PubMed]
- Kiazyk, S.; Ball, T.B. Latent Tuberculosis Infection: An Overview. Can Commun Dis Rep 2017, 43, 62–66. [Google Scholar] [CrossRef]
- UNAIDS 2024 report. The urgency of now: AIDS at a crossroads. Geneva: Joint United Nations Programme on HIV/AIDS; 2024. Licence: CC BY-NC-SA 3.0 IGO. https://www.unaids.org/en/resources/documents/2024/global-aids-update-2024UNAIDS.
- Kitenge, M.K.; Fatti, G.; Eshun-Wilson, I.; Aluko, O.; Nyasulu, P. Prevalence and Trends of Advanced HIV Disease among Antiretroviral Therapy-Naïve and Antiretroviral Therapy-Experienced Patients in South Africa between 2010-2021: A Systematic Review and Meta-Analysis. BMC Infect Dis 2023, 23, 549. [Google Scholar] [CrossRef]
- Abdulrahman, S.A.; Ganasegeran, K.; Rampal, L.; Martins, O.F. HIV Treatment Adherence - A Shared Burden for Patients, Health-Care Providers, and Other Stakeholders. AIDS Rev 2019, 21, 28–39. [Google Scholar] [CrossRef]
- HIV drug resistance: brief report 2024, World Health Organization, 2024. Geneva. Available online: https://iris.who.int/bitstream/handle/10665/376039/9789240086319-eng.pdf?sequence=1 (accessed on March 2025).
- Mandell, L.; Woodhead, M.; Ewig, S.; Torres, A. Respiratory Infections; CRC Press, 2006; ISBN 9780429073366.
- Russell, D.G. Mycobacterium tuberculosis: Here Today, and Here Tomorrow. Nat Rev Mol Cell Biol 2001, 2, 569–577. [Google Scholar] [CrossRef] [PubMed]
- Hosseinian, K.; Gerami, A.; Bral, M.; Venketaraman, V. Mycobacterium tuberculosis-Human Immunodeficiency Virus Infection and the Role of T Cells in Protection. Vaccines (Basel) 2024, 12. [Google Scholar] [CrossRef]
- Stamm, C.E.; Collins, A.C.; Shiloh, M.U. Sensing of Mycobacterium tuberculosis and Consequences to Both Host and Bacillus. Immunol Rev 2015, 264, 204–219. [Google Scholar] [CrossRef] [PubMed]
- Woo, M.; Wood, C.; Kwon, D.; Park, K.-H.P.; Fejer, G.; Delorme, V. Mycobacterium tuberculosis Infection and Innate Responses in a New Model of Lung Alveolar Macrophages. Front Immunol 2018, 9, 438. [Google Scholar] [CrossRef]
- Domingo-Gonzalez, R.; Prince, O.; Cooper, A.; Khader, S.A. Cytokines and Chemokines in Mycobacterium Tuberculosis Infection. Microbiol Spectr 2016, 4. [Google Scholar] [CrossRef]
- Nisa, A.; Kipper, F.C.; Panigrahy, D.; Tiwari, S.; Kupz, A.; Subbian, S. Different Modalities of Host Cell Death and Their Impact on Mycobacterium tuberculosis Infection. Am J Physiol Cell Physiol 2022, 323, C1444–C1474. [Google Scholar] [CrossRef]
- Navasardyan, I.; Miwalian, R.; Petrosyan, A.; Yeganyan, S.; Venketaraman, V. HIV-TB Coinfection: Current Therapeutic Approaches and Drug Interactions. Viruses 2024, 16. [Google Scholar] [CrossRef] [PubMed]
- de Martino, M.; Lodi, L.; Galli, L.; Chiappini, E. Immune Response to Mycobacterium tuberculosis: A Narrative Review. Front Pediatr 2019, 7, 350. [Google Scholar] [CrossRef] [PubMed]
- Cronan, M.R. In the Thick of It: Formation of the Tuberculous Granuloma and Its Effects on Host and Therapeutic Responses. Front Immunol 2022, 13, 820134. [Google Scholar] [CrossRef] [PubMed]
- Ramakrishnan, L. Revisiting the Role of the Granuloma in Tuberculosis. Nat Rev Immunol 2012, 12, 352–366. [Google Scholar] [CrossRef]
- Azevedo-Pereira, J.M.; Pires, D.; Calado, M.; Mandal, M.; Santos-Costa, Q.; Anes, E. HIV/Mtb Co-Infection: From the Amplification of Disease Pathogenesis to an “Emerging Syndemic”. Microorganisms 2023, 11. [Google Scholar] [CrossRef]
- Bares, S.H.; Swindells, S. Latent Tuberculosis and HIV Infection. Curr Infect Dis Rep 2020, 22. [Google Scholar] [CrossRef]
- Diedrich, C.R.; O’Hern, J.; Wilkinson, R.J. HIV-1 and the Mycobacterium tuberculosis Granuloma: A Systematic Review and Meta-Analysis. Tuberculosis (Edinb) 2016, 98, 62–76. [Google Scholar] [CrossRef]
- Jones, R.M.; Adams, K.N.; Eldesouky, H.E.; Sherman, D.R. The Evolving Biology of Mycobacterium tuberculosis Drug Resistance. Front Cell Infect Microbiol 2022, 12, 1027394. [Google Scholar] [CrossRef]
- Sun, W.; Gui, X.; Wu, Z.; Zhang, Y.; Yan, L. Prediction of Drug Resistance Profile of Multidrug-Resistant Mycobacterium tuberculosis (MDR-MTB) Isolates from Newly Diagnosed Case by Whole Genome Sequencing (WGS): A Study from a High Tuberculosis Burden Country. BMC Infect Dis 2022, 22, 499. [Google Scholar] [CrossRef]
- HIV Drug Resistance Report 2021. Geneva: World Health Organization; 2021. Licence: CC BY-NC-SA 3.0 IGO. Available online: https://www.who.int/publications/i/item/9789240038608 (accessed on 14 January 2025).
- Olivença, F.; Nunes, A.; Macedo, R.; Pires, D.; Silveiro, C.; Anes, E.; Miragaia, M.; Gomes, J.P.; Catalão, M.J. Uncovering Beta-Lactam Susceptibility Patterns in Clinical Isolates of Mycobacterium tuberculosis through Whole-Genome Sequencing. Microbiol Spectr 2022, 10, e0067422. [Google Scholar] [CrossRef]
- Pires, D.; Valente, E.; Simões, M.F.; Carmo, N.; Testa, B.; Constantino, L.; Anes, E. Esters of Pyrazinoic Acid Are Active against Pyrazinamide-Resistant Strains of Mycobacterium tuberculosis and Other Naturally Resistant Mycobacteria In Vitro and Ex Vivo within Macrophages. Antimicrob Agents Chemother 2015, 59, 7693–7699. [Google Scholar] [CrossRef] [PubMed]
- Pais, J.P.; Magalhães, M.; Antoniuk, O.; Barbosa, I.; Freire, R.; Pires, D.; Valente, E.; Testa, B.; Anes, E.; Constantino, L. Benzoic Acid Derivatives as Prodrugs for the Treatment of Tuberculosis. Pharmaceuticals (Basel) 2022, 15. [Google Scholar] [CrossRef]
- Centers for Disease Control and Prevention. Treatment for TB Disease. Available online: https://www.cdc.gov/tb/topic/treatment/tbdisease.htm (accessed on 14 January 2025).
- Seung, K.J.; Keshavjee, S.; Rich, M.L. Multidrug-Resistant Tuberculosis and Extensively Drug-Resistant Tuberculosis. Cold Spring Harb Perspect Med 2015, 5, a017863. [Google Scholar] [CrossRef] [PubMed]
- Beloor Suresh, A.; Rosani, A.; Patel, P.; Wadhwa, R. Rifampin; 2025. Available online: https://www.ncbi.nlm.nih.gov/books/NBK557488.
- Koch, A.; Mizrahi, V.; Warner, D.F. The Impact of Drug Resistance on Mycobacterium tuberculosis Physiology: What Can We Learn from Rifampicin? Emerg Microbes Infect 2014, 3, e17. [Google Scholar] [CrossRef] [PubMed]
- Timmins, G.S.; Deretic, V. Mechanisms of Action of Isoniazid. Mol Microbiol 2006, 62, 1220–1227. [Google Scholar] [CrossRef]
- Marrakchi, H.; Lanéelle, M.-A.; Daffé, M. Mycolic Acids: Structures, Biosynthesis, and Beyond. Chem Biol 2014, 21, 67–85. [Google Scholar] [CrossRef]
- Zhang, Y.; Shi, W.; Zhang, W.; Mitchison, D. Mechanisms of Pyrazinamide Action and Resistance. Microbiol Spectr 2014, 2, MGM2-0023–2013. [CrossRef]
- Palomino, J.C.; Martin, A. Drug Resistance Mechanisms in Mycobacterium tuberculosis. Antibiotics (Basel) 2014, 3, 317–340. [Google Scholar] [CrossRef]
- Olivença, F.; Pires, D.; Silveiro, C.; Gama, B.; Holtreman, F.; Anes, E.; Catalão, M.J. Ethambutol and Meropenem/Clavulanate Synergy Promotes Enhanced Extracellular and Intracellular Killing of Mycobacterium tuberculosis. Antimicrob Agents Chemother 2024, 68, e0158623. [Google Scholar] [CrossRef]
- Ennassiri, W.; Jaouhari, S.; Sabouni, R.; Cherki, W.; Charof, R.; Filali-Maltouf, A.; Lahlou, O. Analysis of Isoniazid and Rifampicin Resistance in Mycobacterium tuberculosis Isolates in Morocco Using GenoType® MTBDRplus Assay. J Glob Antimicrob Resist 2018, 12, 197–201. [Google Scholar] [CrossRef]
- Lai, R.P.J.; Meintjes, G.; Wilkinson, R.J. HIV-1 Tuberculosis-Associated Immune Reconstitution Inflammatory Syndrome. Semin Immunopathol 2016, 38, 185–198. [Google Scholar] [CrossRef]
- Sueki, H.; Mizukawa, Y.; Aoyama, Y. Immune Reconstitution Inflammatory Syndrome in Non-HIV Immunosuppressed Patients. J Dermatol 2018, 45, 3–9. [Google Scholar] [CrossRef] [PubMed]
- Manzardo, C.; Guardo, A.C.; Letang, E.; Plana, M.; Gatell, J.M.; Miro, J.M. Opportunistic Infections and Immune Reconstitution Inflammatory Syndrome in HIV-1-Infected Adults in the Combined Antiretroviral Therapy Era: A Comprehensive Review. Expert Rev Anti Infect Ther 2015, 13, 751–767. [Google Scholar] [CrossRef]
- Bruchfeld, J.; Correia-Neves, M.; Kallenius, G. Tuberculosis and HIV Coinfection. Cold Spring Harb Perspect Med 2015, 5. [Google Scholar] [CrossRef]
- Panel on Antiretroviral Guidelines for Adults and Adolescents. Guidelines for the Use of Antiretroviral Agents in Adults and Adolescents With HIV. Department of Health and Human Services. Available online: https://clinicalinfo.hiv.gov/en/guidelines/adult-and-adolescent-arv (accessed on 14 January 2025).
- Niemi, M.; Backman, J.T.; Fromm, M.F.; Neuvonen, P.J.; Kivistö, K.T. Pharmacokinetic Interactions with Rifampicin: Clinical Relevance. Clin Pharmacokinet 2003, 42, 819–850. [Google Scholar] [CrossRef]
- Schutz, C.; Meintjes, G.; Almajid, F.; Wilkinson, R.J.; Pozniak, A. Clinical Management of Tuberculosis and HIV-1 Co-Infection. Eur Respir J 2010, 36, 1460–1481. [Google Scholar] [CrossRef] [PubMed]
- Horne, D.J.; Spitters, C.; Narita, M. Experience with Rifabutin Replacing Rifampin in the Treatment of Tuberculosis. Int J Tuberc Lung Dis 2011, 15, 1485–1489, i. [CrossRef]
- Ignatius, E.H.; Swindells, S. Are We There Yet? Short-Course Regimens in TB and HIV: From Prevention to Treatment of Latent to XDR TB. Curr HIV/AIDS Rep 2020, 17, 589–600. [CrossRef]
- Swindells, S.; Ramchandani, R.; Gupta, A.; Benson, C.A.; Leon-Cruz, J.; Mwelase, N.; Jean Juste, M.A.; Lama, J.R.; Valencia, J.; Omoz-Oarhe, A.; et al. One Month of Rifapentine plus Isoniazid to Prevent HIV-Related Tuberculosis. N Engl J Med 2019, 380, 1001–1011. [Google Scholar] [CrossRef]
- WHO Consolidated Guidelines on Tuberculosis: Tuberculosis Preventive Treatment; World Health Organization, 2020, ISBN 9789240001503.
- Pais, J.P.; Antoniuk, O.; Pires, D.; Delgado, T.; Fortuna, A.; Costa, P.J.; Anes, E.; Constantino, L. Synthesis, Activity, Toxicity, and In Silico Studies of New Antimycobacterial N-Alkyl Nitrobenzamides. Pharmaceuticals (Basel) 2024, 17. [Google Scholar] [CrossRef]
- Carr, W.; Kurbatova, E.; Starks, A.; Goswami, N.; Allen, L.; Winston, C. Interim Guidance: 4-Month Rifapentine-Moxifloxacin Regimen for the Treatment of Drug-Susceptible Pulmonary Tuberculosis - United States, 2022. MMWR Morb Mortal Wkly Rep 2022, 71, 285–289. [Google Scholar] [CrossRef]
- Centers for Disease Control and Prevention. TB Treatment for Persons with HIV. Available online: https://www.cdc.gov/tb/topic/treatment/tbhiv.htm (accessed on 14 January 2025).
- Alexandrova, L.; Zicari, S.; Matyugina, E.; Khandazhinskaya, A.; Smirnova, T.; Andreevskaya, S.; Chernousova, L.; Vanpouille, C.; Kochetkov, S.; Margolis, L. Dual-Targeted Anti-TB/Anti-HIV Heterodimers. Antiviral Res 2017, 145, 175–183. [Google Scholar] [CrossRef]
- Yang, B.; Mukherjee, T.; Radhakrishnan, R.; Paidipally, P.; Ansari, D.; John, S.; Vankayalapati, R.; Tripathi, D.; Yi, G. HIV-Differentiated Metabolite N-Acetyl-L-Alanine Dysregulates Human Natural Killer Cell Responses to Mycobacterium tuberculosis Infection. Int J Mol Sci 2023, 24. [Google Scholar] [CrossRef]
- Zumla, A.; Rao, M.; Wallis, R.S.; Kaufmann, S.H.E.; Rustomjee, R.; Mwaba, P.; Vilaplana, C.; Yeboah-Manu, D.; Chakaya, J.; Ippolito, G.; et al. Host-Directed Therapies for Infectious Diseases: Current Status, Recent Progress, and Future Prospects. Lancet Infect Dis 2016, 16, e47-63. [Google Scholar] [CrossRef] [PubMed]
- Casadevall, A.; Pirofski, L.A. Host-Pathogen Interactions: Basic Concepts of Microbial Commensalism, Colonization, Infection, and Disease. Infect Immun 2000, 68, 6511–6518. [Google Scholar] [CrossRef]
- GBD 2013 Mortality and Causes of Death Collaborators Global, Regional, and National Age-Sex Specific All-Cause and Cause-Specific Mortality for 240 Causes of Death, 1990-2013: A Systematic Analysis for the Global Burden of Disease Study 2013. Lancet 2015, 385, 117–171. [CrossRef] [PubMed]
- Wallis, R.S.; O’Garra, A.; Sher, A.; Wack, A. Host-Directed Immunotherapy of Viral and Bacterial Infections: Past, Present and Future. Nat Rev Immunol 2023, 23, 121–133. [Google Scholar] [CrossRef] [PubMed]
- Shapira, T.; Christofferson, M.; Av-Gay, Y. The Antimicrobial Activity of Innate Host-Directed Therapies: A Systematic Review. Int J Antimicrob Agents 2024, 63, 107138. [Google Scholar] [CrossRef]
- Goletti, D.; Ong, C.W.M.; Friedland, J.S. Host-Directed Therapies: Old and New Approaches for the Treatment of Infections. Int J Infect Dis 2024, 146, 107130. [Google Scholar] [CrossRef]
- Liu, B.; Zhang, W.; Xia, B.; Jing, S.; Du, Y.; Zou, F.; Li, R.; Lu, L.; Chen, S.; Li, Y.; et al. Broadly Neutralizing Antibody-Derived CAR T Cells Reduce Viral Reservoir in Individuals Infected with HIV-1. J Clin Invest 2021, 131. [Google Scholar] [CrossRef]
- Kamyshnyi, A.; Koval, H.; Kobevko, O.; Buchynskyi, M.; Oksenych, V.; Kainov, D.; Lyubomirskaya, K.; Kamyshna, I.; Potters, G.; Moshynets, O. Therapeutic Effectiveness of Interferon-A2b against COVID-19 with Community-Acquired Pneumonia: The Ukrainian Experience. Int J Mol Sci 2023, 24. [Google Scholar] [CrossRef]
- Muir, A.J.; Arora, S.; Everson, G.; Flisiak, R.; George, J.; Ghalib, R.; Gordon, S.C.; Gray, T.; Greenbloom, S.; Hassanein, T.; et al. A Randomized Phase 2b Study of Peginterferon Lambda-1a for the Treatment of Chronic HCV Infection. J Hepatol 2014, 61, 1238–1246. [Google Scholar] [CrossRef]
- Tam, A.R.; Zhang, R.R.; Lung, K.-C.; Liu, R.; Leung, K.-Y.; Liu, D.; Fan, Y.; Lu, L.; Lam, A.H.-Y.; Chung, T.W.-H.; et al. Early Treatment of High-Risk Hospitalized Coronavirus Disease 2019 (COVID-19) Patients with a Combination of Interferon Beta-1b and Remdesivir: A Phase 2 Open-Label Randomized Controlled Trial. Clin Infect Dis 2023, 76, e216–e226. [Google Scholar] [CrossRef]
- Mapamba, D.A.; Sauli, E.; Mrema, L.; Lalashowi, J.; Magombola, D.; Buza, J.; Olomi, W.; Wallis, R.S.; Ntinginya, N.E. Impact of N-Acetyl Cysteine (NAC) on Tuberculosis (TB) Patients-A Systematic Review. Antioxidants (Basel) 2022, 11. [Google Scholar] [CrossRef] [PubMed]
- Vita, S.; Rosati, S.; Ascoli Bartoli, T.; Beccacece, A.; D’Abramo, A.; Mariano, A.; Scorzolini, L.; Goletti, D.; Nicastri, E. Monoclonal Antibodies for Pre- and Postexposure Prophylaxis of COVID-19: Review of the Literature. Pathogens 2022, 11. [Google Scholar] [CrossRef]
- Raghuveer, T.S.; Zackula, R. Nirsevimab for Prevention of RSV Hospitalizations in Infants. N Engl J Med 2024, 390, 1152. [Google Scholar] [CrossRef]
- Ferraccioli, G.; Gremese, E.; Goletti, D.; Petrone, L.; Cantini, F.; Ugel, S.; Canè, S.; Bronte, V. Immune-Guided Therapy of COVID-19. Cancer Immunol Res 2022, 10, 384–402. [Google Scholar] [CrossRef]
- Goulenok, T.; Gaudemer, A.; Rouzaud, D.; Chauveheid, M.-P.; Alexandra, J.-F.; Sacre, K.; Papo, T. Infliximab to Treat Severe Paradoxical Reaction in HIV-Negative Tuberculous Meningoencephalitis. Neurology 2022, 98, 118–119. [Google Scholar] [CrossRef]
- BARNARD, C. Tuberculous Meningitis; Cortisone Treatment as an Adjunct to the Antibiotics; the Effect on the Clinical Features and the Cerebrospinal Fluid. S Afr Med J 1953, 27, 219–220. [Google Scholar] [PubMed]
- Chandra, H.; Rahman, A.; Yadav, P.; Maurya, G.; Kumar Shukla, S. Effect of Adjunct Vitamin D Treatment in Vitamin D Deficient Pulmonary Tuberculosis Patients: A Randomized, Double Blind, Active Controlled Clinical Trial. Indian J Tuberc 2024, 71, 170–178. [Google Scholar] [CrossRef]
- Dutta, N.K.; Bruiners, N.; Pinn, M.L.; Zimmerman, M.D.; Prideaux, B.; Dartois, V.; Gennaro, M.L.; Karakousis, P.C. Statin Adjunctive Therapy Shortens the Duration of TB Treatment in Mice. J Antimicrob Chemother 2016, 71, 1570–1577. [Google Scholar] [CrossRef] [PubMed]
- Mai, N.T.H.; Dobbs, N.; Phu, N.H.; Colas, R.A.; Thao, L.T.P.; Thuong, N.T.T.; Nghia, H.D.T.; Hanh, N.H.H.; Hang, N.T.; Heemskerk, A.D.; et al. A Randomised Double Blind Placebo Controlled Phase 2 Trial of Adjunctive Aspirin for Tuberculous Meningitis in HIV-Uninfected Adults. Elife 2018, 7. [Google Scholar] [CrossRef]
- Bourigault, M.-L.; Vacher, R.; Rose, S.; Olleros, M.L.; Janssens, J.-P.; Quesniaux, V.F.; Garcia, I. Tumor Necrosis Factor Neutralization Combined with Chemotherapy Enhances Mycobacterium tuberculosis Clearance and Reduces Lung Pathology. Am J Clin Exp Immunol 2013, 2, 124–134. [Google Scholar]
- Wallis, R.S.; Kyambadde, P.; Johnson, J.L.; Horter, L.; Kittle, R.; Pohle, M.; Ducar, C.; Millard, M.; Mayanja-Kizza, H.; Whalen, C.; et al. A Study of the Safety, Immunology, Virology, and Microbiology of Adjunctive Etanercept in HIV-1-Associated Tuberculosis. AIDS 2004, 18, 257–264. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Sun, J. Vitamin D, Vitamin D Receptor, and Macroautophagy in Inflammation and Infection. Discov Med 2011, 11, 325–335. [Google Scholar] [PubMed]
- Mily, A.; Rekha, R.S.; Kamal, S.M.M.; Akhtar, E.; Sarker, P.; Rahim, Z.; Gudmundsson, G.H.; Agerberth, B.; Raqib, R. Oral Intake of Phenylbutyrate with or without Vitamin D3 Upregulates the Cathelicidin LL-37 in Human Macrophages: A Dose Finding Study for Treatment of Tuberculosis. BMC Pulm Med 2013, 13, 23. [Google Scholar] [CrossRef]
- Mily, A.; Rekha, R.S.; Kamal, S.M.M.; Arifuzzaman, A.S.M.; Rahim, Z.; Khan, L.; Haq, M.A.; Zaman, K.; Bergman, P.; Brighenti, S.; et al. Significant Effects of Oral Phenylbutyrate and Vitamin D3 Adjunctive Therapy in Pulmonary Tuberculosis: A Randomized Controlled Trial. PLoS One 2015, 10, e0138340. [Google Scholar] [CrossRef] [PubMed]
- Wejse, C.; Gomes, V.F.; Rabna, P.; Gustafson, P.; Aaby, P.; Lisse, I.M.; Andersen, P.L.; Glerup, H.; Sodemann, M. Vitamin D as Supplementary Treatment for Tuberculosis: A Double-Blind, Randomized, Placebo-Controlled Trial. Am J Respir Crit Care Med 2009, 179, 843–850. [Google Scholar] [CrossRef]
- Martineau, A.R.; Timms, P.M.; Bothamley, G.H.; Hanifa, Y.; Islam, K.; Claxton, A.P.; Packe, G.E.; Moore-Gillon, J.C.; Darmalingam, M.; Davidson, R.N.; et al. High-Dose Vitamin D (3) during Intensive-Phase Antimicrobial Treatment of Pulmonary Tuberculosis: A Double-Blind Randomised Controlled Trial. Lancet 2011, 377, 242–250. [Google Scholar] [CrossRef]
- Cubillos-Angulo, J.M.; Nogueira, B.M.F.; Arriaga, M.B.; Barreto-Duarte, B.; Araújo-Pereira, M.; Fernandes, C.D.; Vinhaes, C.L.; Villalva-Serra, K.; Nunes, V.M.; Miguez-Pinto, J.P.; et al. Host-Directed Therapies in Pulmonary Tuberculosis: Updates on Anti-Inflammatory Drugs. Front Med (Lausanne) 2022, 9, 970408. [Google Scholar] [CrossRef]
- Marzo, E.; Vilaplana, C.; Tapia, G.; Diaz, J.; Garcia, V.; Cardona, P.-J. Damaging Role of Neutrophilic Infiltration in a Mouse Model of Progressive Tuberculosis. Tuberculosis (Edinb) 2014, 94, 55–64. [Google Scholar] [CrossRef]
- Byrne, S.T.; Denkin, S.M.; Zhang, Y. Aspirin and Ibuprofen Enhance Pyrazinamide Treatment of Murine Tuberculosis. J Antimicrob Chemother 2007, 59, 313–316. [Google Scholar] [CrossRef]
- Vilaplana, C.; Marzo, E.; Tapia, G.; Diaz, J.; Garcia, V.; Cardona, P.-J. Ibuprofen Therapy Resulted in Significantly Decreased Tissue Bacillary Loads and Increased Survival in a New Murine Experimental Model of Active Tuberculosis. J Infect Dis 2013, 208, 199–202. [Google Scholar] [CrossRef]
- Shim, D.; Kim, H.; Shin, S.J. Mycobacterium tuberculosis Infection-Driven Foamy Macrophages and Their Implications in Tuberculosis Control as Targets for Host-Directed Therapy. Front Immunol 2020, 11, 910. [Google Scholar] [CrossRef] [PubMed]
- Parihar, S.P.; Guler, R.; Khutlang, R.; Lang, D.M.; Hurdayal, R.; Mhlanga, M.M.; Suzuki, H.; Marais, A.D.; Brombacher, F. Statin Therapy Reduces the Mycobacterium tuberculosis Burden in Human Macrophages and in Mice by Enhancing Autophagy and Phagosome Maturation. J Infect Dis 2014, 209, 754–763. [Google Scholar] [CrossRef] [PubMed]
- Skerry, C.; Pinn, M.L.; Bruiners, N.; Pine, R.; Gennaro, M.L.; Karakousis, P.C. Simvastatin Increases the in Vivo Activity of the First-Line Tuberculosis Regimen. J Antimicrob Chemother 2014, 69, 2453–2457. [Google Scholar] [CrossRef] [PubMed]
- Wallis, R.S.; Hafner, R. Advancing Host-Directed Therapy for Tuberculosis. Nat Rev Immunol 2015, 15, 255–263. [Google Scholar] [CrossRef]
- Kang, Y.A.; Choi, N.-K.; Seong, J.-M.; Heo, E.Y.; Koo, B.K.; Hwang, S.-S.; Park, B.-J.; Yim, J.-J.; Lee, C.-H. The Effects of Statin Use on the Development of Tuberculosis among Patients with Diabetes Mellitus. Int J Tuberc Lung Dis 2014, 18, 717–724. [Google Scholar] [CrossRef]
- Pandey, A.K.; Sassetti, C.M. Mycobacterial Persistence Requires the Utilization of Host Cholesterol. Proc Natl Acad Sci U S A 2008, 105, 4376–4380. [Google Scholar] [CrossRef]
- Guerra-De-Blas, P.D.C.; Bobadilla-Del-Valle, M.; Sada-Ovalle, I.; Estrada-García, I.; Torres-González, P.; López-Saavedra, A.; Guzmán-Beltrán, S.; Ponce-de-León, A.; Sifuentes-Osornio, J. Simvastatin Enhances the Immune Response Against Mycobacterium tuberculosis. Front Microbiol 2019, 10, 2097. [Google Scholar] [CrossRef]
- Rodrigues Díez, R.; Rodrigues-Díez, R.; Lavoz, C.; Rayego-Mateos, S.; Civantos, E.; Rodríguez-Vita, J.; Mezzano, S.; Ortiz, A.; Egido, J.; Ruiz-Ortega, M. Statins Inhibit Angiotensin II/Smad Pathway and Related Vascular Fibrosis, by a TGF-β-Independent Process. PLoS One 2010, 5, e14145. [Google Scholar] [CrossRef]
- Ma, Y.-X.; Li, W.-H.; Xie, Q. Rosuvastatin Inhibits TGF-Beta1 Expression and Alleviates Myocardial Fibrosis in Diabetic Rats. Pharmazie 2013, 68, 355–358. [Google Scholar]
- Sojka, D.; Šnebergerová, P.; Robbertse, L. Protease Inhibition-An Established Strategy to Combat Infectious Diseases. Int J Mol Sci 2021, 22. [Google Scholar] [CrossRef]
- Drag, M.; Salvesen, G.S. Emerging Principles in Protease-Based Drug Discovery. Nat Rev Drug Discov 2010, 9, 690–701. [Google Scholar] [CrossRef]
- Bond, J.S. Proteases: History, Discovery, and Roles in Health and Disease. J Biol Chem 2019, 294, 1643–1651. [Google Scholar] [CrossRef] [PubMed]
- Rawlings, N.D.; Bateman, A. How to Use the MEROPS Database and Website to Help Understand Peptidase Specificity. Protein Sci 2021, 30, 83–92. [Google Scholar] [CrossRef] [PubMed]
- Barrett, A.J.; Rawling, N.D.; Woessner, J.F. Handbook of Proteolytic Enzymes Second Edition Volume 1 Aspartic and Metallo Peptidases Editor Biographies Xxi.
- Renslo, A.R.; McKerrow, J.H. Drug Discovery and Development for Neglected Parasitic Diseases. Nat Chem Biol 2006, 2, 701–710. [Google Scholar] [CrossRef] [PubMed]
- McKerrow, J.H.; Caffrey, C.; Kelly, B.; Loke, P.; Sajid, M. Proteases in Parasitic Diseases. Annu Rev Pathol 2006, 1, 497–536. [Google Scholar] [CrossRef]
- Kitchen, V.S.; Skinner, C.; Ariyoshi, K.; Lane, E.A.; Duncan, I.B.; Burckhardt, J.; Burger, H.U.; Bragman, K.; Pinching, A.J.; Weber, J.N. Safety and Activity of Saquinavir in HIV Infection. Lancet 1995, 345, 952–955. [Google Scholar] [CrossRef]
- Kosalaraksa, P.; Ananworanich, J.; Puthanakit, T.; Pinyakorn, S.; Lumbiganon, P.; Chuanjaroen, T.; Chobkarjing, U.; Phanuphak, P.; Pancharoen, C.; Bunupuradah, T.; et al. Long-Term Lopinavir/Ritonavir Monotherapy in HIV-Infected Children. Pediatr Infect Dis J 2013, 32, 350–353. [Google Scholar] [CrossRef]
- Ghosh, A.K.; Osswald, H.L.; Prato, G. Recent Progress in the Development of HIV-1 Protease Inhibitors for the Treatment of HIV/AIDS. J Med Chem 2016, 59, 5172–5208. [Google Scholar] [CrossRef]
- McKerrow, J.H. Update on Drug Development Targeting Parasite Cysteine Proteases. PLoS Negl Trop Dis 2018, 12, e0005850. [Google Scholar] [CrossRef]
- Richardson, P.G. Update on Proteasome Inhibitors in Multiple Myeloma. Clin Adv Hematol Oncol 2014, 12, 179–181. [Google Scholar]
- Bibo-Verdugo, B.; Jiang, Z.; Caffrey, C.R.; O’Donoghue, A.J. Targeting Proteasomes in Infectious Organisms to Combat Disease. FEBS J 2017, 284, 1503–1517. [Google Scholar] [CrossRef] [PubMed]
- Hilgenfeld, R. From SARS to MERS: Crystallographic Studies on Coronaviral Proteases Enable Antiviral Drug Design. FEBS J 2014, 281, 4085–4096. [Google Scholar] [CrossRef] [PubMed]
- Rut, W.; Lv, Z.; Zmudzinski, M.; Patchett, S.; Nayak, D.; Snipas, S.J.; El Oualid, F.; Huang, T.T.; Bekes, M.; Drag, M.; et al. Activity Profiling and Crystal Structures of Inhibitor-Bound SARS-CoV-2 Papain-like Protease: A Framework for Anti-COVID-19 Drug Design. Sci Adv 2020, 6. [Google Scholar] [CrossRef]
- Freitas, B.T.; Durie, I.A.; Murray, J.; Longo, J.E.; Miller, H.C.; Crich, D.; Hogan, R.J.; Tripp, R.A.; Pegan, S.D. Characterization and Noncovalent Inhibition of the Deubiquitinase and DeISGylase Activity of SARS-CoV-2 Papain-Like Protease. ACS Infect Dis 2020, 6, 2099–2109. [Google Scholar] [CrossRef]
- Shin, D.; Mukherjee, R.; Grewe, D.; Bojkova, D.; Baek, K.; Bhattacharya, A.; Schulz, L.; Widera, M.; Mehdipour, A.R.; Tascher, G.; et al. Papain-like Protease Regulates SARS-CoV-2 Viral Spread and Innate Immunity. Nature 2020, 587, 657–662. [Google Scholar] [CrossRef] [PubMed]
- Lupoli, T.J.; Vaubourgeix, J.; Burns-Huang, K.; Gold, B. Targeting the Proteostasis Network for Mycobacterial Drug Discovery. ACS Infect Dis 2018, 4, 478–498. [Google Scholar] [CrossRef]
- Pires, D.; Marques, J.; Pombo, J.P.; Carmo, N.; Bettencourt, P.; Neyrolles, O.; Lugo-Villarino, G.; Anes, E. Role of Cathepsins in Mycobacterium tuberculosis Survival in Human Macrophages. Sci Rep 2016, 6, 32247. [Google Scholar] [CrossRef]
- Pires, D.; Bernard, E.M.; Pombo, J.P.; Carmo, N.; Fialho, C.; Gutierrez, M.G.; Bettencourt, P.; Anes, E. Mycobacterium tuberculosis Modulates MiR-106b-5p to Control Cathepsin S Expression Resulting in Higher Pathogen Survival and Poor T-Cell Activation. Front Immunol 2017, 8, 1819. [Google Scholar] [CrossRef]
- Nsanzabana, C.; Rosenthal, P.J. In Vitro Activity of Antiretroviral Drugs against Plasmodium Falciparum. Antimicrob Agents Chemother 2011, 55, 5073–5077. [Google Scholar] [CrossRef]
- Castilho, V.V.S.; Gonçalves, K.C.S.; Rebello, K.M.; Baptista, L.P.R.; Sangenito, L.S.; Santos, H.L.C.; Branquinha, M.H.; Santos, A.L.S.; Menna-Barreto, R.F.S.; Guimarães, A.C.; et al. Docking Simulation between HIV Peptidase Inhibitors and Trypanosoma Cruzi Aspartyl Peptidase. BMC Res Notes 2018, 11, 825. [Google Scholar] [CrossRef]
- Savarino, A. Expanding the Frontiers of Existing Antiviral Drugs: Possible Effects of HIV-1 Protease Inhibitors against SARS and Avian Influenza. J Clin Virol 2005, 34, 170–178. [Google Scholar] [CrossRef]
- Pires, D.; Valente, S.; Calado, M.; Mandal, M.; Azevedo-Pereira, J.M.; Anes, E. Repurposing Saquinavir for Host-Directed Therapy to Control Mycobacterium tuberculosis Infection. Front Immunol 2021, 12, 647728. [Google Scholar] [CrossRef] [PubMed]
- Conus, S.; Simon, H.-U. Cathepsins and Their Involvement in Immune Responses. Swiss Med Wkly 2010, 140, w13042. [Google Scholar] [CrossRef] [PubMed]
- Soualhine, H.; Deghmane, A.-E.; Sun, J.; Mak, K.; Talal, A.; Av-Gay, Y.; Hmama, Z. Mycobacterium Bovis Bacillus Calmette-Guérin Secreting Active Cathepsin S Stimulates Expression of Mature MHC Class II Molecules and Antigen Presentation in Human Macrophages. J Immunol 2007, 179, 5137–5145. [Google Scholar] [CrossRef]
- Beers, C.; Burich, A.; Kleijmeer, M.J.; Griffith, J.M.; Wong, P.; Rudensky, A.Y. Cathepsin S Controls MHC Class II-Mediated Antigen Presentation by Epithelial Cells in Vivo. J Immunol 2005, 174, 1205–1212. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.-L.; Chang, W.-S.W.; Cheung, C.H.A.; Lin, C.-C.; Huang, C.-C.; Yang, Y.-N.; Kuo, C.-P.; Kuo, C.-C.; Chang, Y.-H.; Liu, K.-J.; et al. Targeting Cathepsin S Induces Tumor Cell Autophagy via the EGFR-ERK Signaling Pathway. Cancer Lett 2012, 317, 89–98. [Google Scholar] [CrossRef]
- Espert, L.; Beaumelle, B.; Vergne, I. Autophagy in Mycobacterium tuberculosis and HIV Infections. Front Cell Infect Microbiol 2015, 5, 49. [Google Scholar] [CrossRef]
- Lim, J.; Park, H.; Heisler, J.; Maculins, T.; Roose-Girma, M.; Xu, M.; Mckenzie, B.; van Lookeren Campagne, M.; Newton, K.; Murthy, A. Autophagy Regulates Inflammatory Programmed Cell Death via Turnover of RHIM-Domain Proteins. Elife 2019, 8. [Google Scholar] [CrossRef]
- Pires, D.; Mandal, M.; Pinho, J.; Catalão, M.J.; Almeida, A.J.; Azevedo-Pereira, J.M.; Gaspar, M.M.; Anes, E. Liposomal Delivery of Saquinavir to Macrophages Overcomes Cathepsin Blockade by Mycobacterium tuberculosis and Helps Control the Phagosomal Replicative Niches. Int J Mol Sci 2023, 24. [Google Scholar] [CrossRef]
- Turk, V.; Stoka, V.; Vasiljeva, O.; Renko, M.; Sun, T.; Turk, B.; Turk, D. Cysteine Cathepsins: From Structure, Function and Regulation to New Frontiers. Biochim Biophys Acta 2012, 1824, 68–88. [Google Scholar] [CrossRef]
- Russell, D.G.; Vanderven, B.C.; Glennie, S.; Mwandumba, H.; Heyderman, R.S. The Macrophage Marches on Its Phagosome: Dynamic Assays of Phagosome Function. Nat Rev Immunol 2009, 9, 594–600. [Google Scholar] [CrossRef]
- Russell, D.G. New Ways to Arrest Phagosome Maturation. Nat Cell Biol 2007, 9, 357–359. [Google Scholar] [CrossRef]
- Welin, A.; Raffetseder, J.; Eklund, D.; Stendahl, O.; Lerm, M. Importance of Phagosomal Functionality for Growth Restriction of Mycobacterium tuberculosis in Primary Human Macrophages. J Innate Immun 2011, 3, 508–518. [Google Scholar] [CrossRef] [PubMed]
- Magister, S.; Kos, J. Cystatins in Immune System. J Cancer 2013, 4, 45–56. [Google Scholar] [CrossRef] [PubMed]
- Ochieng, J.; Chaudhuri, G. Cystatin Superfamily. J Health Care Poor Underserved 2010, 21, 51–70. [Google Scholar] [CrossRef] [PubMed]
- Lautwein, A.; Burster, T.; Lennon-Duménil, A.-M.; Overkleeft, H.S.; Weber, E.; Kalbacher, H.; Driessen, C. Inflammatory Stimuli Recruit Cathepsin Activity to Late Endosomal Compartments in Human Dendritic Cells. Eur J Immunol 2002, 32, 3348–3357. [Google Scholar] [CrossRef]
- Colbert, J.D.; Matthews, S.P.; Kos, J.; Watts, C. Internalization of Exogenous Cystatin F Supresses Cysteine Proteases and Induces the Accumulation of Single-Chain Cathepsin L by Multiple Mechanisms. J Biol Chem 2011, 286, 42082–42090. [Google Scholar] [CrossRef]
- Ogrinc, T.; Dolenc, I.; Ritonja, A.; Turk, V. Purification of the Complex of Cathepsin L and the MHC Class II-Associated Invariant Chain Fragment from Human Kidney. FEBS Lett 1993, 336, 555–559. [Google Scholar] [CrossRef]
- Anes, E.; Azevedo-Pereira, J.M.; Pires, D. Cathepsins and Their Endogenous Inhibitors in Host Defense During Mycobacterium tuberculosis and HIV Infection. Front Immunol 2021, 12, 726984. [Google Scholar] [CrossRef]
- Pires, D.; Calado, M.; Velez, T.; Mandal, M.; Catalão, M.J.; Neyrolles, O.; Lugo-Villarino, G.; Vérollet, C.; Azevedo-Pereira, J.M.; Anes, E. Modulation of Cystatin C in Human Macrophages Improves Anti-Mycobacterial Immune Responses to Mycobacterium tuberculosis Infection and Coinfection With HIV. Front Immunol 2021, 12, 742822. [Google Scholar] [CrossRef]
- Pires, D.; Mandal, M.; Matos, A.I.; Peres, C.; Catalão, M.J.; Azevedo-Pereira, J.M.; Satchi-Fainaro, R.; Florindo, H.F.; Anes, E. Development of Chitosan Particles Loaded with SiRNA for Cystatin C to Control Intracellular Drug-Resistant Mycobacterium tuberculosis. Antibiotics (Basel) 2023, 12. [Google Scholar] [CrossRef] [PubMed]
- Mandal, M.; Pires, D.; Catalão, M.J.; Azevedo-Pereira, J.M.; Anes, E. Modulation of Cystatin F in Human Macrophages Impacts Cathepsin-Driven Killing of Multidrug-Resistant Mycobacterium tuberculosis. Microorganisms 2023, 11. [Google Scholar] [CrossRef] [PubMed]
- Ni, J.; Fernandez, M.A.; Danielsson, L.; Chillakuru, R.A.; Zhang, J.; Grubb, A.; Su, J.; Gentz, R.; Abrahamson, M. Cystatin F Is a Glycosylated Human Low Molecular Weight Cysteine Proteinase Inhibitor. J Biol Chem 1998, 273, 24797–24804. [Google Scholar] [CrossRef]
- Langerholc, T.; Zavasnik-Bergant, V.; Turk, B.; Turk, V.; Abrahamson, M.; Kos, J. Inhibitory Properties of Cystatin F and Its Localization in U937 Promonocyte Cells. FEBS J 2005, 272, 1535–1545. [Google Scholar] [CrossRef] [PubMed]
- Mandal, M.; Pires, D.; Calado, M.; Azevedo-Pereira, J.M.; Anes, E. Cystatin F Depletion in Mycobacterium tuberculosis-Infected Macrophages Improves Cathepsin C/Granzyme B-Driven Cytotoxic Effects on HIV-Infected Cells during Coinfection. Int J Mol Sci 2024, 25. [Google Scholar] [CrossRef]
- Cohen, L.A.; Light, R.W. Tuberculous Pleural Effusion. Turk Thorac J 2015, 16, 1–9. [Google Scholar] [CrossRef]
- Werle, B.; Sauckel, K.; Nathanson, C.-M.; Bjarnadottir, M.; Spiess, E.; Ebert, W.; Abrahamson, M. Cystatins C, E/M and F in Human Pleural Fluids of Patients with Neoplastic and Inflammatory Lung Disorders. Biol Chem 2003, 384, 281–287. [Google Scholar] [CrossRef]
Figure 1.
Immune response in TB and during HIV coinfection. Alveolar macrophages attempt to contain the infection by phagocytizing Mtb, however, the pathogen is able to establish intracellular survival niches within endosomes. The innate immune response is executed by coordinating the activity of macrophages, NK cells, and DC, which secrete an array of effector cytokines (eg. TNF-α, IFN-γ, IL-1) as well as various chemokines. This process recruits CD4+ and CD8+ T cells to release their specific cytokines, such as IFN-γ, which activates infected macrophages to a more bactericidal state. This, in turn, creates a concerted immune cascade that leads to granuloma formation. Apoptosis of cells infected with Mtb is promoted by TNF-α following activation by IFN-γ or by the cytotoxic activity of CTLs. This process contributes to the control of the infection in a latent form within the host. However, the introduction of HIV, results in a systemic immunosuppression that is characterized by a dramatic loss of functional CD4+ T cells by apoptosis. This depletion of CD4+ T cell leads to the disruption of granuloma architecture and stability, this enabling Mtb to evade immune surveillance helping progression to active TB infection.
Figure 1.
Immune response in TB and during HIV coinfection. Alveolar macrophages attempt to contain the infection by phagocytizing Mtb, however, the pathogen is able to establish intracellular survival niches within endosomes. The innate immune response is executed by coordinating the activity of macrophages, NK cells, and DC, which secrete an array of effector cytokines (eg. TNF-α, IFN-γ, IL-1) as well as various chemokines. This process recruits CD4+ and CD8+ T cells to release their specific cytokines, such as IFN-γ, which activates infected macrophages to a more bactericidal state. This, in turn, creates a concerted immune cascade that leads to granuloma formation. Apoptosis of cells infected with Mtb is promoted by TNF-α following activation by IFN-γ or by the cytotoxic activity of CTLs. This process contributes to the control of the infection in a latent form within the host. However, the introduction of HIV, results in a systemic immunosuppression that is characterized by a dramatic loss of functional CD4+ T cells by apoptosis. This depletion of CD4+ T cell leads to the disruption of granuloma architecture and stability, this enabling Mtb to evade immune surveillance helping progression to active TB infection.
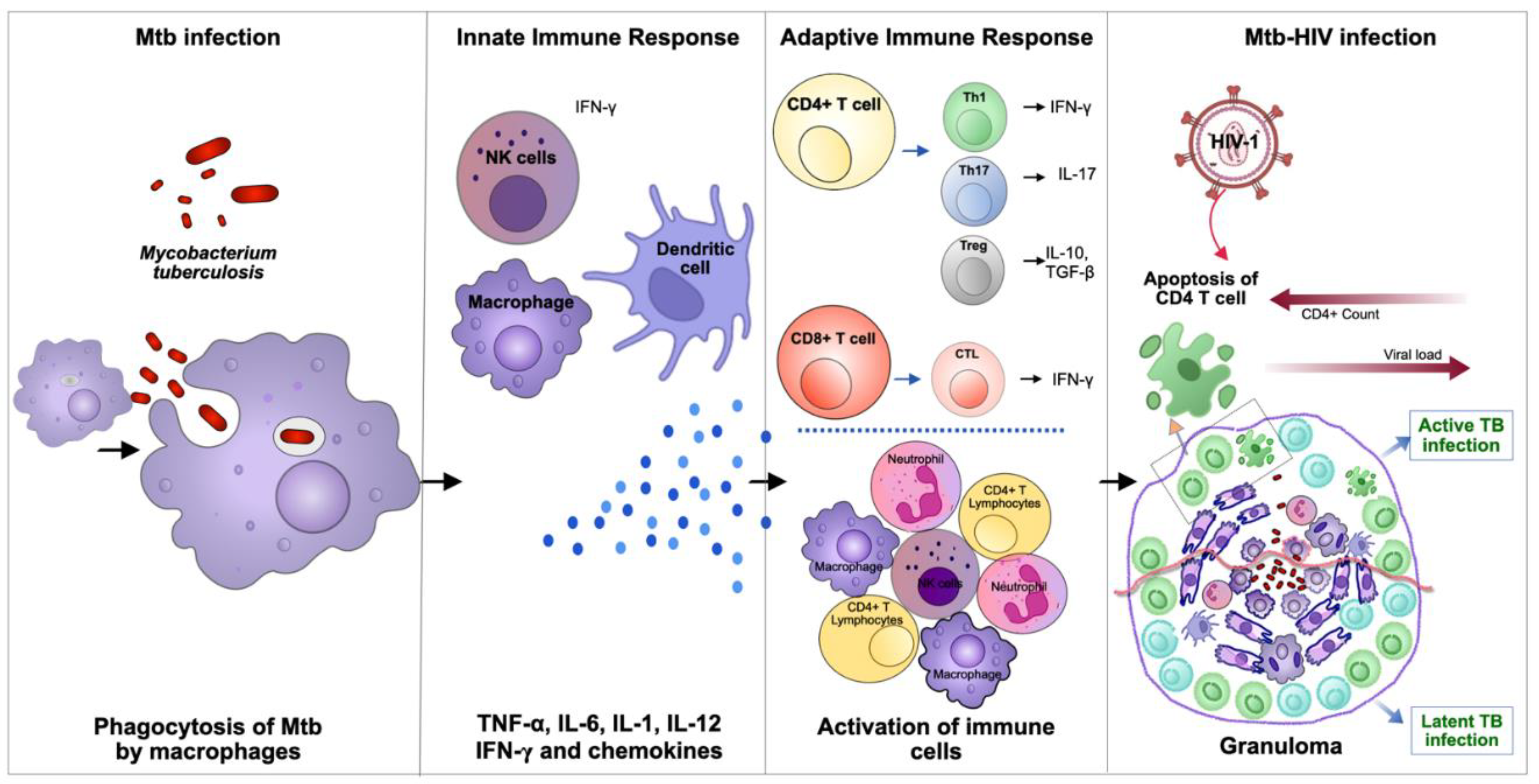
Figure 2.
Host-directed therapies (HDT) against Mycobacterium tuberculosis. A. Main HDT used to improve outcome in Mtb. (1) Drugs/compounds in HDT can affect granuloma integrity to augment access for drugs (2) A few agents in HDT have the ability to suppress proinflammatory reactions that contribute to inflammation and tissue damage at active stage of Mtb infection (3) The administration of monoclonal antibodies appears to be a new concept in HDT that has been shown to inhibit Mtb replication (4) HDT agents like Doxycycline, Statins and Metformin modulate cell-mediated immunity, including antigen-specific T cell responses (5) Some HDT such as vitamin D can also augment autophagy in infected cells. B. Development of new host-directed strategies against Mtb. The investigation of SQV as a repurposed drug and targeting cystatin C (CstC) and cystatin F (CstF) in human macrophages may represent a promising host-directed strategy to control Mtb infection and a new potential adjuvant therapy for TB.
Figure 2.
Host-directed therapies (HDT) against Mycobacterium tuberculosis. A. Main HDT used to improve outcome in Mtb. (1) Drugs/compounds in HDT can affect granuloma integrity to augment access for drugs (2) A few agents in HDT have the ability to suppress proinflammatory reactions that contribute to inflammation and tissue damage at active stage of Mtb infection (3) The administration of monoclonal antibodies appears to be a new concept in HDT that has been shown to inhibit Mtb replication (4) HDT agents like Doxycycline, Statins and Metformin modulate cell-mediated immunity, including antigen-specific T cell responses (5) Some HDT such as vitamin D can also augment autophagy in infected cells. B. Development of new host-directed strategies against Mtb. The investigation of SQV as a repurposed drug and targeting cystatin C (CstC) and cystatin F (CstF) in human macrophages may represent a promising host-directed strategy to control Mtb infection and a new potential adjuvant therapy for TB.
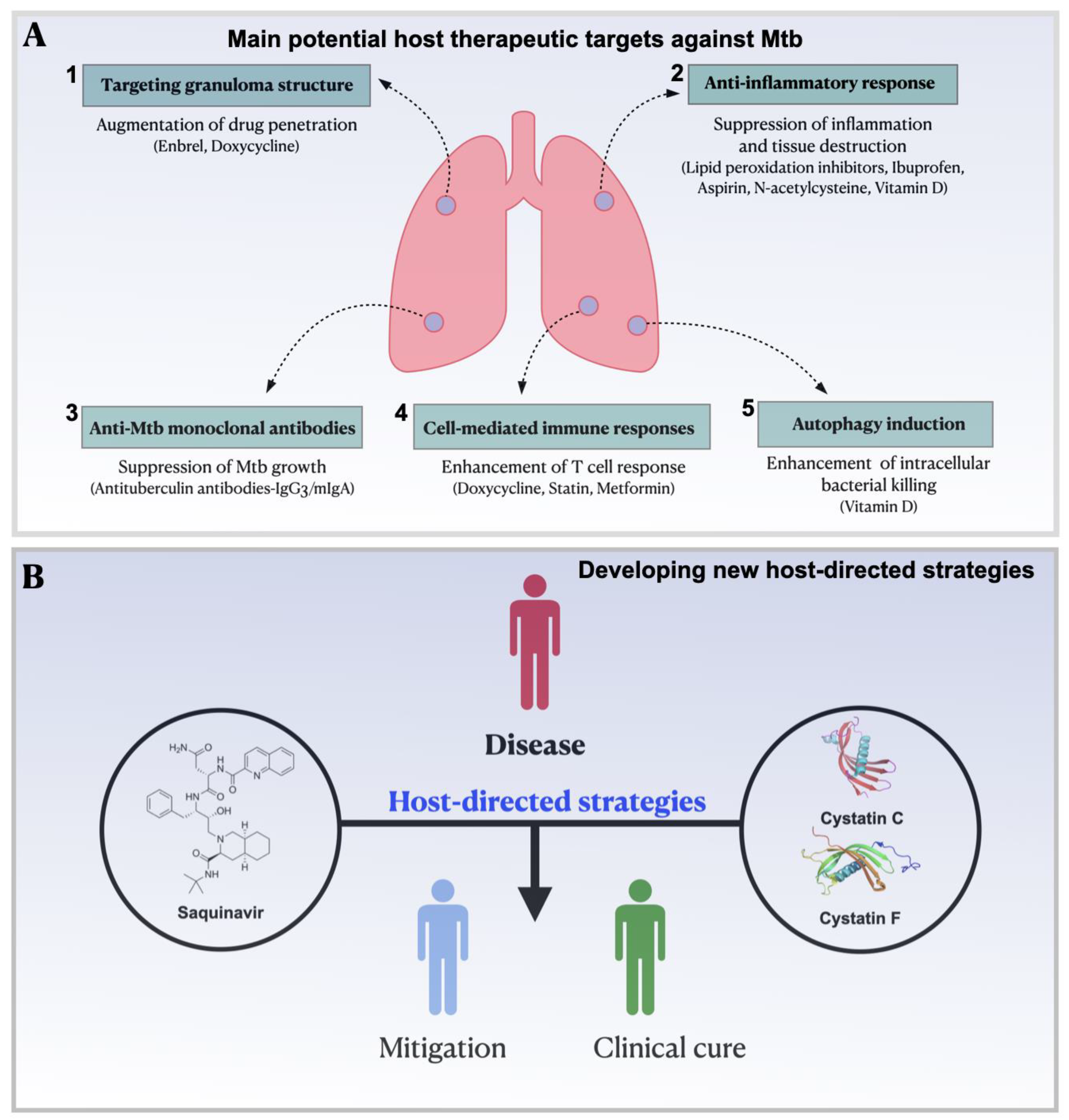
Figure 3.
CstF depletion in Mtb-infected macrophages improves the proteolytic activity of cathepsins L, B, and S, significantly impacting the intracellular survival of the pathogen. Interference in CstF expression could restore the basal levels of proteolytic activity that were detected prior to infection with Mtb.
Figure 3.
CstF depletion in Mtb-infected macrophages improves the proteolytic activity of cathepsins L, B, and S, significantly impacting the intracellular survival of the pathogen. Interference in CstF expression could restore the basal levels of proteolytic activity that were detected prior to infection with Mtb.
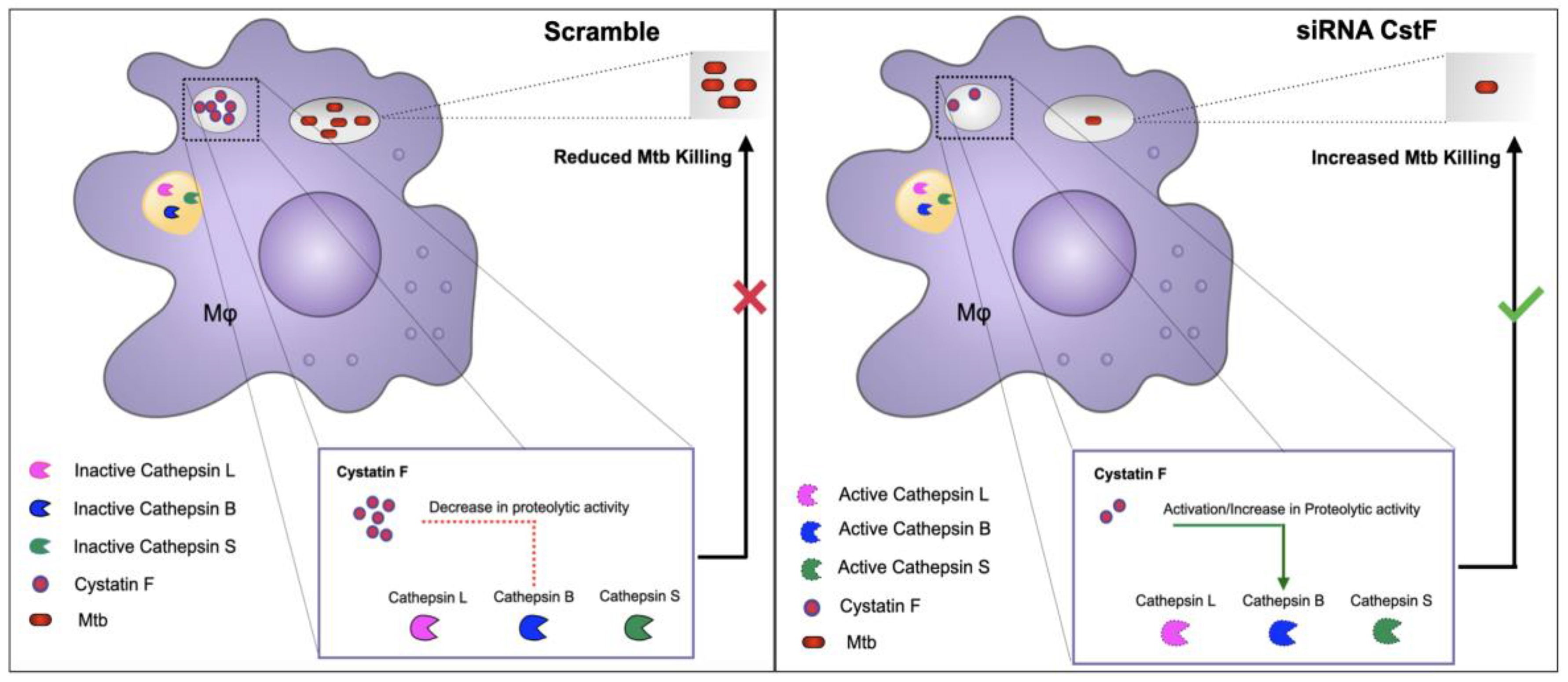
Figure 4.
Impact of CstF depletion from Mtb-infected macrophages on the cytotoxic activity of lymphocytes. During the coinfection of Mtb-infected macrophages with lymphocytes infected with HIV, CstF depletion enhances cathepsin C enzymatic activity, which in turn activates granzyme B. This process results in a notable decrease in the viral loads. Adapted from [139].
Figure 4.
Impact of CstF depletion from Mtb-infected macrophages on the cytotoxic activity of lymphocytes. During the coinfection of Mtb-infected macrophages with lymphocytes infected with HIV, CstF depletion enhances cathepsin C enzymatic activity, which in turn activates granzyme B. This process results in a notable decrease in the viral loads. Adapted from [139].
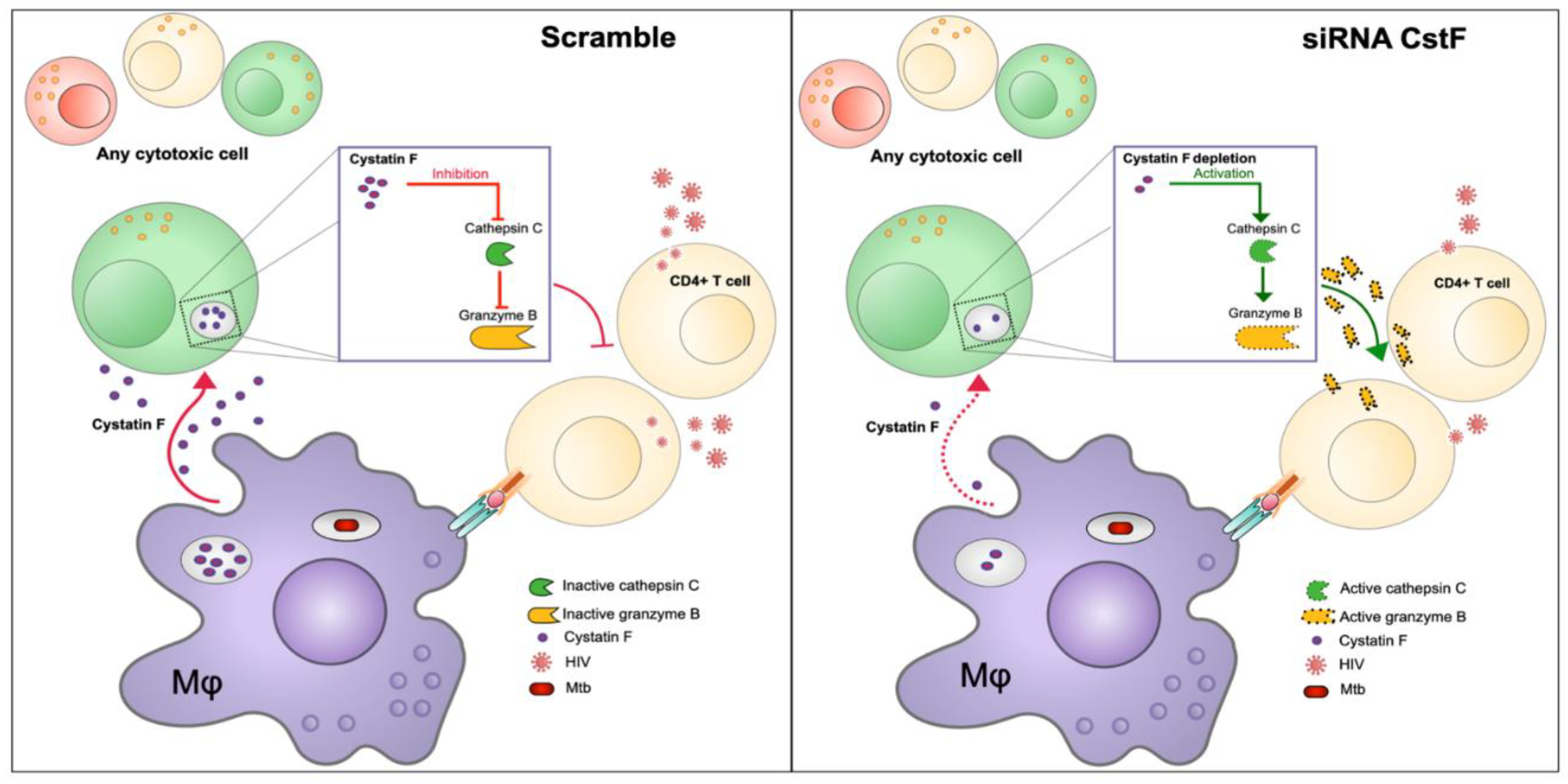
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



