
Submitted:
06 March 2025
Posted:
07 March 2025
You are already at the latest version
Abstract
Atherosclerotic cardiovascular disease (ASCVD) has been screened for decades using the traditional lipid profile, with by far the most emphasis on low-density lipoprotein-cholesterol (LDL). Even though more recent studies have suggested that there might be more to atherosclerosis than LDL, with inflammation and immune processes being some of the possible culprits, solid recommendations still haven’t been made regarding the potential use of other biomarkers. Most of the research has focused on Lipoprotein(a) [Lp(a)], an independent cardiovascular (CV) risk factor, with involvement in the progression of atherosclerosis, yet dramatically underused in clinical practice. This review aims to highlight the reasons for the lack of Lp(a) screening in clinical practice and to address these potential limitations. The study also reviews current screening and treatment methods and explores the hypothesis that proprotein convertase subtilisin/kexin type 9 inhibitors (PCSK9-i) provide CV benefits due to a reduction in Lp(a), rather than LDL. A comprehensive review of the literature was conducted to assess the role of Lp(a) in cardiovascular disease risk, compare its predictive value to traditional lipid markers, and evaluate different measurement techniques, including mass spectrometry and immunoassays. Additionally, the review examined both current treatment strategies and emerging therapeutic approaches, such as gene-silencing therapies, that may offer new possibilities for managing elevated Lp(a) levels. One in four clinicians does not routinely check Lp(a) levels, which proves a lack of awareness amongst them. The reasons for that are implying that the cost is too high and that available treatments are scarce. The traditional lipid profile, including LDL, high-density lipoprotein (HDL) and triglycerides, continues to be the gold standard for CV risk assessment. However, Lp(a) has emerged as a potential biomarker for ASCVD, particularly in patients with high baseline levels or in patients who continue to develop CV events, despite having controlled LDL levels. One limitation of using Lp(a) in clinical practice is the significant variability in apo(a) sizes, which results from the presence of multiple isoforms determined by the number of kringle domains. This structural diversity poses challenges in standardizing measurement methods, affecting the accuracy and comparability of results. While statins have minimal impact on Lp(a), PCSK9-i lower its levels by 20-25%, although this class is not prescribed primarily for this reason. Lastly, gene-silencing therapies, which achieve the most reduction in Lp(a) levels, are still in phase III trials, and there is still a need to examine whether the reduction translates into CV benefits. Currently, there is more than one challenge that needs to be addressed when it comes to the use of Lp(a) as a reliable biomarker. Among them, the most mentioned in the literature are the lack of standardization and the absence of data to support a treatment. This should not discourage further research, because ASCVD’s complexity requires a more tailored approach. Current lipid-lowering therapy still fails in a minority of cases, as evidenced by new-onset cardiovascular events in patients with well-controlled LDL levels. There is a need for future interventional studies to assess whether the reduction in Lp(a) by PCSK9-i really translates into CV benefits, independent of LDL.
Keywords:
lipoprotein(a)
; LDL-cholesterol
; cardiovascular disease
; atherosclerosis
; PCSK9-i
; ASO
; SiRNA
1. Introduction
In recent years, cardiovascular disease (CVD) has remained the leading cause of mortality worldwide, with atherosclerosis being one of the culprits [1]. Traditional risk factors, such as diabetes, dyslipidemia, hypertension have been proven to contribute to the development of atherosclerotic plaque. However, the focus has started shifting towards new, promising risk factors, including adhesion molecules, pro-inflammatory molecules, oxidized phospholipids and Lp(a) [2,3].
The traditional lipid profile has been used for decades as an important tool for screening ASCVD. However, recent studies have linked atherosclerosis to inflammation and immune processes, suggesting that LDL is no longer the sole driver for atherosclerosis development. Furthermore, the deposition of cholesterol particles is now thought to be an active phenomenon, rather than a passive one [3].
There are many limitations to the use of Lp(a) as a potential biomarker. Amongst them, the lack of current validated treatments is a major one. Studies evaluating the efficacy and safety of antisense oligonucleotides (ASO) are still ongoing, with results expected to be available in the coming years. However, even if we surpass this limitation, we still have a subtle but significant challenge: the myriad of apo(a) isoforms, which directly influence the structure of Lp(a) and make current screening tests less reliable due to a lack of standardization [4]. This highlights the importance of carefully evaluating whether Lp(a) has been measured using the most reliable methods in trials where lowering Lp(a) was the primary objective.
The genetic inheritance of Lp(a) level and the absence of its fluctuation with lifestyle or current treatments diminished efforts toward its future research, and instead, attention shifted back to LDL, exploiting it even more. The initial reduction in the mean LDL determined by alirocumab in the Odyssey Outcomes trial waned after 4 months and even further by the 48-month mark, but its positive CV outcomes persisted, suggesting that other mechanisms influenced by PCSK9-i underlie these outcomes [5,6].
The purpose of this review is to emphasize the urgent need for a standardized method in determining Lp(a), given the variety of apo(a) isoforms, which in turn discourages screening in clinical practice [7,8]. It also highlights the insufficient research into the relationship between PCSK9-i and Lp(a) [5]. By improving our knowledge and guiding research in this direction, Lp(a) could move closer to a clinical implementation alongside the traditional lipid profile.
2. Material and Methods
2.1. Research Strategy
We conducted a search through international databases such as PubMed, Google Scholar, ResearchGate, Scopus and Web of Science, with an emphasis on randomized clinical trials, cohort studies, case-control studies, systematic reviews and meta-analyses. Keywords such as ‘lipoprotein(a)’, ‘CV risk’, ‘PCSK9-i’, ‘antisense oligonucleotides’ and ‘atherosclerosis’ were used to refine the search process and to ensure adequate results. Ongoing clinical trials were also reviewed, highlighting the methodologies used for Lp(a) serum determination and the study outcomes. Last but not least, based on the results, we outlined future directions that could serve as a starting point for translating theoretical findings to practical application. This review follows PRISMA (Preferred Reporting Items for Systematic Reviews and Meta-Analyses) guidelines.
2.2. Inclusion and Exclusion Criteria
The selection of studies was based on predefined inclusion and exclusion criteria. Studies were included if they were randomized controlled trials, cohort studies, case-control studies, meta-analyses, or systematic reviews published in the last 5 years in English. Only studies focusing on lipoprotein(a) measurement, screening methods, its role in cardiovascular disease, and potential therapies were considered.
Exclusion criteria included non-peer-reviewed sources, animal studies, duplicate publications, and those with incomplete data. Studies older than the specified period that did not provide additional information were also excluded.
2.3. Study Selection
The initial search yielded 2,196 results. Following a triage based on titles and abstracts, 56 studies were selected. Despite containing the most recent data, some relevant information was still missing. To address this, the timeframe was extended by five years (2014–2024), resulting in the inclusion of 15 additional studies. However, there were consistent gaps regarding certain screening methods. To comprehensively understand these limitations, six more studies were added, with the oldest dating back to 2000.
3. Results and Discussion
3.1. Lipoprotein(a): Structure and Pathophysiology
Lipoprotein(a) stands out among lipoproteins due to its unique structure, consisting of apo-B100, an LDL-like particle, which covalently binds to apo(a), a plasminogen-like particle, through a disulfide bond [4,9]. Therefore, Lp(a) combines the inflammation and atherogenesis from LDL, with the prothrombotic risk of plasminogen [10,11,12,13]. Its physiological role is yet to be discovered, but since it resembles plasminogen, it might act as an antifibrinolytic factor [4,14]. Apo(a) is composed of kringle domains (KIV and KV), which vary in size and exist in numerous isoforms. This variability in apo(a) structure ultimately leads to the size differences observed in Lp(a) [4,9].
Genetic traits are the most recognized factors that play a role in the variability of Lp(a), accounting for almost 90% of it [15]. This is also the reason why Lp(a) levels remain fairly constant throughout an individual’s life, but vary significantly between individuals. Individuals of African descent have higher serum concentrations compared to Caucasians [16]. Kamal Awad et al. published a study in 2024, stating that there is a relationship between Lp(a) and other CV risk factors. The highest individual variability of Lp(a), defined as >10 mg/dL, was observed in individuals with high LDL, those undergoing statin treatment and those with a history of CVD. Lp(a) concentration is mostly regulated on the production site. To make the most impact, this factor must be considered in the development of future treatments. The primary target for medication should be the production site, specifically the liver [14,17,18].
One of the key features of Lp(a) is increasing the expression of pro-inflammatory cytokines, such as Interleukin-6 (IL-6), Interleukin-8 (IL-8), Tumor necrosis factor-α (TNF-α) and adhesion molecules, like E-selectin, Vascular cell adhesion molecule-1 (VCAM-1) and P-selectin. This promotes the chemoattraction of immune cells, thereby amplifying inflammation and ultimately contributing to the progression of atherosclerosis [19,20,21]. Lp(a) also carries approximately 70% of plasma oxidized phospholipids, which directly influence plaque composition, making it more unstable and, consequently, more prone to rupture. Patients with elevated Lp(a) levels are more likely to present with acute rather than chronic coronary syndrome [3,21,22,23].
A Mendelian randomization study published in 2016 by Emdin CA et al. found that by genetically lowering Lp(a), the risk for heart disease is reduced. There was a risk reduction by 37% for aortic stenosis, 31% for peripheral artery disease, 29% for coronary heart disease, 17% for heart failure and 13% for stroke. The study included data from 112,338 participants and had some significant limitations, including the reliance on patients’ reports regarding their past medical history rather than verifying medical records and the relatively low variability regarding ancestry– the majority of the subjects had European descent [24].
Data from two Danish studies, which included a total of 98,097 subjects, were analyzed by Pia R. Kamstrup et al., and the results suggested that the opposite is also true – high Lp(a) levels increase the risk of heart failure by 18%, mostly because of the effect of Lp(a) on other cardiovascular diseases like myocardial infarction and valvular disease. The diagnosis of heart failure was obtained from the patient’s history, without conducting new echocardiographic studies to evaluate ventricular function at the time of the enrollment [25].
A study of 2527 subjects with established CVD, published in 2019 by Christian M. Madsen et al., evaluated the risk of major cardiovascular events (MACE) during a median follow-up of 5 years. MACE rates ranged from 29 per 1000 person-years for Lp(a)<10 mg/dL to 54 per 1000 person-years for Lp(a)≥100 mg/dL. Also, the study showed that in secondary prevention, a 20% or 40% reduction in MACE could be achieved by an Lp(a) reduction of 50 mg/dL and 99 mg/dL, respectively. More than one type of assay was used in this study to determine serum Lp(a) levels, and although results were calibrated, this still predisposes to inconsistent measurements [26,27].
A meta-analysis of 29,069 patients, including 8064 women, with repeat Lp(a) measurements, found a linear association between both baseline and on-statin Lp(a) levels and cardiovascular disease risk, with increased risk observed at Lp(a) levels of 30 mg/dL or higher for baseline and 50 mg/dL or higher for on-statin Lp(a) [28].
3.2. Screening
For many decades, the traditional lipid profile has been the number one tool used for screening atherosclerotic disease. Recent studies have also linked atherosclerosis to inflammation and immune processes, suggesting that a high LDL-cholesterol level is no longer the only driver for developing plaques, and the accumulation of lipids in the arterial wall is not at all a passive phenomenon [23,29,30,31]. These findings opened a new path of research, trying to find other markers that would further refine the screening process [2,11]. Findings were promising, but many of these markers, like CRP (C-reactive protein), Fibrinogen, Serum Amyloid S, IL-6, IL-8, TNF-α, adhesion molecules, or microparticles, lack specificity, and/or from an economic standpoint, the screening would be unfeasible[32,33,34,35,36,37].
Serum LDL cholesterol determination is ubiquitously the most commonly used laboratory test for estimating cardiovascular risk related to dyslipidemia. Numerous studies have shown a linear relationship between LDL cholesterol levels and atherosclerotic disease risk, with lowering LDL also reducing cardiovascular risk [38,39,40]. A meta-analysis published in 2010 by the Cholesterol Treatment Trialists (CTT) Collaboration demonstrated that lowering LDL cholesterol through statin therapy significantly reduces the incidence of major vascular events, including coronary death and non-fatal myocardial infarction, by approximately 15% per 10 mmol/L reduction in LDL cholesterol, without any significant side effects [41].
Associated to LDL-cholesterol, the traditional lipid profile also includes HDL (high-density lipoprotein)-cholesterol and triglycerides for a more precise assessment of a patient’s metabolic profile. The advantage of this traditional lipid profile, besides its proven efficacy, lies in its cost-effectiveness and the relative simplicity of the available methods, compared with Lp(a) determination, which is more expensive and less standardized [42]. A study conducted by the National Institutes of Health in collaboration with the International Federation of Clinical Chemistry evaluated a proposed reference material to standardize Lp(a) measurements across various methods. While calibration using the reference material improved uniformity in control samples, significant variability in measurements persisted due to heterogeneity in apo(a) size, undermining the accuracy of risk assessments for coronary artery disease[43]. Even though we might be able to face this challenge, regarding the variability in apo(a) size, we still have other obstacles ahead, like the difference between mass-based (mg/dL) and molar-based (nmol/L) assays. Conversion between these units remains imprecise, with commercial kits still widely using less reliable mass-based measurements [44].
Various ELISA methods have been developed for measuring Lp(a), each with unique features and limitations. These methods use antibodies that target different Lp(a) components, with sensitivity and detection range varying depending on the antibody used. However, most are susceptible to inaccuracies due to the isoform variability of apo(a) [45].
Both immunoturbidimetry and nephelometry use turbidity measurements to determine Lp(a), with slight differences. While turbidimetry measures turbidity by analyzing light transmission, nephelometry relies on light scattering. Specific reagents can be used to improve the accuracy of these methods. Although they provide broad measurement ranges, they are less sensitive to the variability in apo(a) size [46].
Radial immunodiffusion (RID), or the Mancini method, is a straightforward immunoassay that measures Lp(a) by observing the precipitate formed during the antigen-antibody reaction. Although it can detect Lp(a) concentrations as low as 1.5 mg/dl, it cannot assess the size variability of apo(a). A double-antibody radioimmunoassay, which improves sensitivity, has been developed but is more time-consuming, complex, and prone to higher variability [45].
Electrophoresis techniques, including immunofixation electrophoresis, counterimmunoelectrophoresis, and rocket immunoelectrophoresis, are commonly used for the analysis of Lp(a) concentrations in serum, but they have the same limitations as those previously listed. While these methods offer sensitivity and allow for quantification, they are unable to measure the size variability of Lp(a) or its heterogeneity. Liquid chromatography-mass spectrometry (LC-MS/MS) has emerged as a promising tool for Lp(a) quantification, providing accurate results and better standardization compared to the traditional methods [47].
Commercial immunoassays are widely used for high-throughput measurement of Lp(a), but their accuracy is compromised by sensitivity to apo(a) isoform size variations, which can lead to inconsistent results. Monoclonal antibody-based ELISA assays are more reliable for assessing the relationship between Lp(a) and cardiovascular disease risk, as they are less affected by isoform size variability, providing more consistent and accurate measurements. Denka-based assays, developed by Denka Seiken Co. Ltd, are considered the least affected by isoforms among commercially available options due to their use of multiple calibrators that cover a broad range of apo(a) isoform distributions, thus ensuring more accurate Lp(a) measurements [45].
Szarek et al. compared three methods for measuring lipoprotein(a): two immunoassay-based methods (IA-mass and IA-molar) and one mass spectrometry (MS)-based method. Samples were collected, frozen, and analyzed using standardized procedures. Immunoassay methods used antibodies and calibration standards, while the MS method was isoform-independent and directly quantified specific peptides from apolipoprotein(a). The study found that all three methods were similarly effective at predicting cardiovascular risk, specifically the likelihood of MACE, in patients with recent acute coronary syndrome receiving high-intensity or maximum-tolerated statins. The MS-based method correlated better with the molar immunoassay than with the mass-based immunoassay, but differences between the molar methods highlighted a lack of full equivalence. Despite this, all three tests showed comparable utility for assessing cardiovascular risk and evaluating treatment benefits with alirocumab [48].
In a multicenter cross-sectional epidemiological study conducted by Nissen S et al., 48,135 patients across 48 countries with documented ASCVD were analyzed. Given that only almost one in ten (13.9%) of the subjects had prior Lp(a) measurements, it can be inferred that screening for Lp(a) in individuals with established atherosclerosis is insufficient. This suggests that screening in otherwise healthy individuals may be even less frequent, although further studies would be needed to confirm this. Furthermore, the study revealed that over 25% of those who had their Lp(a) levels measured exhibited values above 50 mg/dL (or 125 nmol/L), a threshold that may be linked to the etiology of their ASCVD [7].
A survey done by The European Atherosclerosis Society (EAS) Lipid Clinic network had the aim of finding out when and how lipoprotein(a) is measured in clinical practice. Almost one quarter (24.5%) of the practitioners included in the survey stated that they do not routinely check Lp(a), the reason being that the cost is too high and the fact that there is a lack of treatment options [8]. This observation should prompt action to improve the availability of Lp(a) measurement and to further research the potential use of PCSK9 inhibitors for their proven Lp(a)-lowering effect, at least until the use of nucleic acid–based gene silencing therapies that specifically target the apo(a) gene transcripts becomes readily available [6,49,50,51,52].
In 2019 ESC’s guidelines for managing dyslipidemias, there is a class IIa level C recommendation for screening Lp(a) at least one time in every individual’s life, comparing the risk of very elevated Lp(a) with familial hypercholesterolemia. This recommendation is especially directed towards those with early onset CVD, recurrent CVD despite optimal medical treatment, family history of premature CVD or elevated Lp(a), aortic stenosis, or a very high cardiovascular risk according to Framingham or ESC Systematic Coronary Risk Evaluation (SCORE). If the recommendations for the target population are reasonably standardized, the guidelines for follow-up are less so. There should be a more aggressive control of traditional risk factors (e.g. LDL, smoking, diabetes). However, no specific recommendations address the elevated Lp(a) levels [53,54].
3.3. Treatment Options
Some treatment options like niacin have, indeed, inconclusive evidence. In a meta-analysis conducted by Sahebkar A. et al., the effect of extended-release nicotinic acid (ER niacin) on plasma Lp(a) concentrations was assessed using data from 14 randomized placebo-controlled trials, including 9013 subjects. The treatment was associated with a significant reduction in Lp(a) levels, with a weighted mean difference of -22.90% (95% CI: -27.32, -18.48, p<0.001). The reduction was consistent across different dose ranges (<2000 mg/day and ≥2000 mg/day) [55]. Schwartz et al. published a review in 2022 that concluded that the lowering effect of niacin on Lp(a) has none or a slight benefit at best [56].
Statins, as potent inhibitors of HMG-CoA reductase, effectively reduce LDL-c levels and cardiovascular risk in both primary and secondary prevention, with additional anti-inflammatory properties observed in high-potency statins. However, their impact on Lp(a) appears minimal, with studies indicating a slight increase in Lp(a) levels, particularly with high-intensity statin therapy. Despite this, the substantial benefits in LDL-c reduction and inflammation control outweigh the negligible Lp(a) increase, emphasizing the continued importance of statins as a cornerstone therapy for ASCVD risk management, while highlighting the need for targeted therapies to address Lp(a) hypercholesterolemia [57]. A systematic review and meta-analysis of 39 randomized trials, published in 2022 (24,448 participants) assessed the impact of statins on Lp(a) levels compared to placebo. The pooled data indicated minimal absolute changes (1.1 mg/dL, 95% CI: 0.5–1.6, P<0.0001) and no significant percentage changes (0.1%, 95% CI: −3.6% to 4.0%, P=0.95). These findings suggest that statin therapy does not significantly alter Lp(a) levels or influence Lp(a)-associated cardiovascular risk in patients at risk for CVD [58].
Lipoprotein apheresis (LA), a procedure similar to dialysis, had been shown to be effective in lowering LDL-cholesterol and Lp(a), which translates into many CV benefits, like improving endothelial dysfunction, coronary artery blood flow, and lowering low-grade inflammation, some of these effects lowering the drive for atherosclerotic plaques to form [59]. Víšek et al. followed 14 familial hypercholesterolemia (FH) patients for 15 years, with the aim to evaluate the efficacy of LA in preventing cardiovascular disease progression. The results showed promising results, with LA significantly reducing cholesterol levels, inflammation markers, and endothelial dysfunction in patients with familial hypercholesterolemia (FH), both immediately after procedures and over time. These findings suggest that sustained LA treatment enhances lipid profiles and vascular health, offering promising cardiovascular benefits for FH patients. One of the studies’ key limitation is that all patients were also treated with high-dose statins, ezetimibe, and PCSK9 inhibitors, which could have contributed to the observed benefits, making it difficult to isolate the effects of LA alone [60]. The recommendation between different guidelines varies, and the reason why it is not that frequently used in practice, in spite of the proven benefits, is caused by the unavailability and the complexity of the method [61,62,63].
The Odyssey Outcomes trial was a multicenter, double-blind, randomized trial that evaluated 18,924 patients with a recent acute coronary syndrome and elevated LDL cholesterol levels despite high-intensity statin therapy. Participants received either alirocumab or placebo every two weeks, targeting an LDL cholesterol level of 25–50 mg/dL, with the primary endpoint being a composite of major adverse cardiovascular events such as death from coronary heart disease, myocardial infarction, ischemic stroke, or unstable angina requiring hospitalization. The study highlighted that alirocumab, a PCSK9 inhibitor, significantly reduced both mortality and cardiovascular events, particularly in patients with inadequately controlled LDL cholesterol levels. However, after an initial decrease in serum LDL, to as low as 40 mg/dL at the 4th month mark, LDL again increased to 66 mg/dL by the 48th month, a level which, by the ESC guidelines, is considered uncontrolled for high-risk patients (target is <55 mg/dL for high-risk patients), yet cardiovascular benefits persisted. If LDL cholesterol, the primary therapeutic target of alirocumab, increased again while cardiovascular benefits persisted, this suggests that the drug may confer its advantages through additional mechanisms beyond LDL cholesterol reduction. Alirocumab also demonstrated a 20-25% reduction in Lp(a) levels (samples were analyzed with an immunoturbidimetric assay), potentially contributing to these effects. Additional research is essential to fully understand the pathways responsible for these extended cardiovascular benefits. The inclusion criteria are a major limitation of the study -only patients with a history of acute coronary syndromes were included, meaning that the results might not apply to the general population, for example, young patients with high Lp(a) levels and no other risk factor [5,53].
Nucleic acid-based gene silencing, involving antisense oligonucleotide (ASO) or short-interfering ribonucleic acid (siRNA) strategies, represents a new and more advanced approach for suppressing the intracellular production of PCSK9 and apo(a) [52,64]. Antisense oligonucleotides are usually administered subcutaneously and act on the cell nucleus by inhibiting the transcription of a specific protein, such as PCSK9 and apo(a), which further decreases the amount of LDL-cholesterol or Lp (a) produced by the liver. One example of such a drug is Pelacarsen, which targets the production of apo(a) [52,65]. SiRNA therapy uses conjugated nucleic acids that act by suppressing a specific gene expression, interfering with the translation and synthesis of new proteins. The difference between ASO and siRNA lies in their structure, allowing siRNA therapy to be administered less often. Inclisiran is an siRNA drug that targets PCSK9, while Olpasiran is also part of the same class but targets apo(a) of the LPA gene [52,66,67].
A phase 2B trial evaluated the impact of pelacarsen, a hepatocyte-targeted antisense oligonucleotide, on lowering Lp(a) in 286 patients with cardiovascular disease and elevated baseline Lp(a) levels (≥60 mg/dL). Participants received pelacarsen in varying regimens or placebo for 6–12 months, with Lp(a) levels assessed at baseline, 6 months, and 16 weeks post-treatment, using an isoform-independent assay, which involves plasma being added to monoclonal antibody LPA4-conjugated magnetic beads in the presence of proline and epsilon-aminocaproic acid. Pelacarsen demonstrated dose-dependent efficacy, with mean Lp(a)-C reductions ranging from 29% to 67% compared to a minimal 2% decrease in the placebo group. An advanced assay allowed precise Lp(a)-C measurements with high sensitivity and broad applicability across population Lp(a) levels. Temporal trends revealed consistent reductions in Lp(a), though levels partially rebounded after discontinuation of therapy [68]. The findings suggest that pelacarsen offers a promising approach to managing elevated Lp(a)-mediated cardiovascular risks, with further research needed to explore long-term outcomes. A phase 3, double-blind, randomized, placebo-controlled, multicenter trial known as Lp(a) HORIZON is currently underway to evaluate the safety and effectiveness of pelacarsen [69].
In ORION studies (1, 3, 9, 10) inclisiran consistently lowered Lp(a) by approximately 15–26%, with greater reductions in two-dose regimens and long-term treatment. These reductions were more pronounced compared to placebo, though the effect on Lp(a) was less pronounced than its effects on LDL-C and apoB levels. These findings align with those from studies evaluating PCSK9 inhibitors, such as the ODYSSEY OUTCOMES trial (alirocumab) and the FOURIER study (evolocumab). Although inclisiran's mechanism of action differs, these drugs ultimately target the same enzyme. This highlights inclisiran’s potential in addressing Lp(a)-mediated cardiovascular risks, especially when used alongside other lipid-lowering therapies [5,70,71].
In a randomized, placebo-controlled trial involving patients with atherosclerotic cardiovascular disease and elevated Lp(a) (>150 nmol/L), olpasiran significantly reduced Lp(a) concentrations across all doses. At 36 weeks, placebo-adjusted reductions ranged from −68.5% with the 10 mg dose every 12 weeks to −100% with the 225 mg dose every 12 weeks. Additionally, 67% to 100% of patients achieved Lp(a) levels of <125 nmol/L depending on the dose and regimen. The trial included only patients with documented ASCVD and a serum level of Lp(a) of at least 150 nmol/L, or approximately 70 mg/dL, so the results might not apply to the general population or subjects with a slight increase in Lp(a). Also, there is no mention of what specific method was used for determining the concentration of Lp(a), which is an important factor given the various isoforms of apo(a) that can affect the measurement [72].
Phase 1 clinical trials, using Roche immunoassay, demonstrated the efficacy and safety of lepodisiran, enabling its progression to phase 2 trials [73]. Lepodisiran showed excellent tolerability and produced significant, dose-dependent reductions in serum lipoprotein(a) levels (a reduction in mean value from 41% in the 4 mg-group, to 97% in the 608 mg-group), with effects sustained over a prolonged duration (mean Lp(a) value decreased with 94% at day 337). These promising results highlight the potential of lepodisiran and support its further clinical evaluation, to see whether the decrease in Lp(a) translates into lowering CVD risk [74].
While adverse effects associated with Lp(a)-lowering therapies are rare and typically limited to injection-site reactions, the need for more convenient and cost-effective treatments has driven the development of oral alternatives. One such therapy is muvalaplin, a novel oral small-molecule inhibitor. Muvalaplin disrupts the interaction between apolipoprotein B-100 and apolipoprotein(a), a critical step in the assembly of Lp(a), thereby significantly reducing circulating Lp(a) concentrations [75]. In the KRAKEN trial, muvalaplin demonstrated remarkable efficacy, with mean Lp(a) levels reduced by up to 85.8% in participants receiving the highest therapeutic dose (240 mg daily). These reductions are considered clinically significant and have positioned muvalaplin as a promising candidate for addressing elevated Lp(a). However, several limitations of this trial warrant attention. First, the study employed commercial assays for Lp(a) measurement, which may underestimate the true magnitude of Lp(a) lowering. Additionally, the trial's short duration (12 weeks) limits insights into long-term safety and efficacy. Importantly, it remains to be determined whether the observed Lp(a) reductions will translate into meaningful reductions in cardiovascular (CV) risk. Muvalaplin is currently undergoing phase 3 clinical trials to further evaluate its safety, efficacy, and potential role in reducing cardiovascular events. If successful, it could provide a significant advancement in the management of elevated Lp(a), particularly for patients seeking non-injectable treatment options [76].

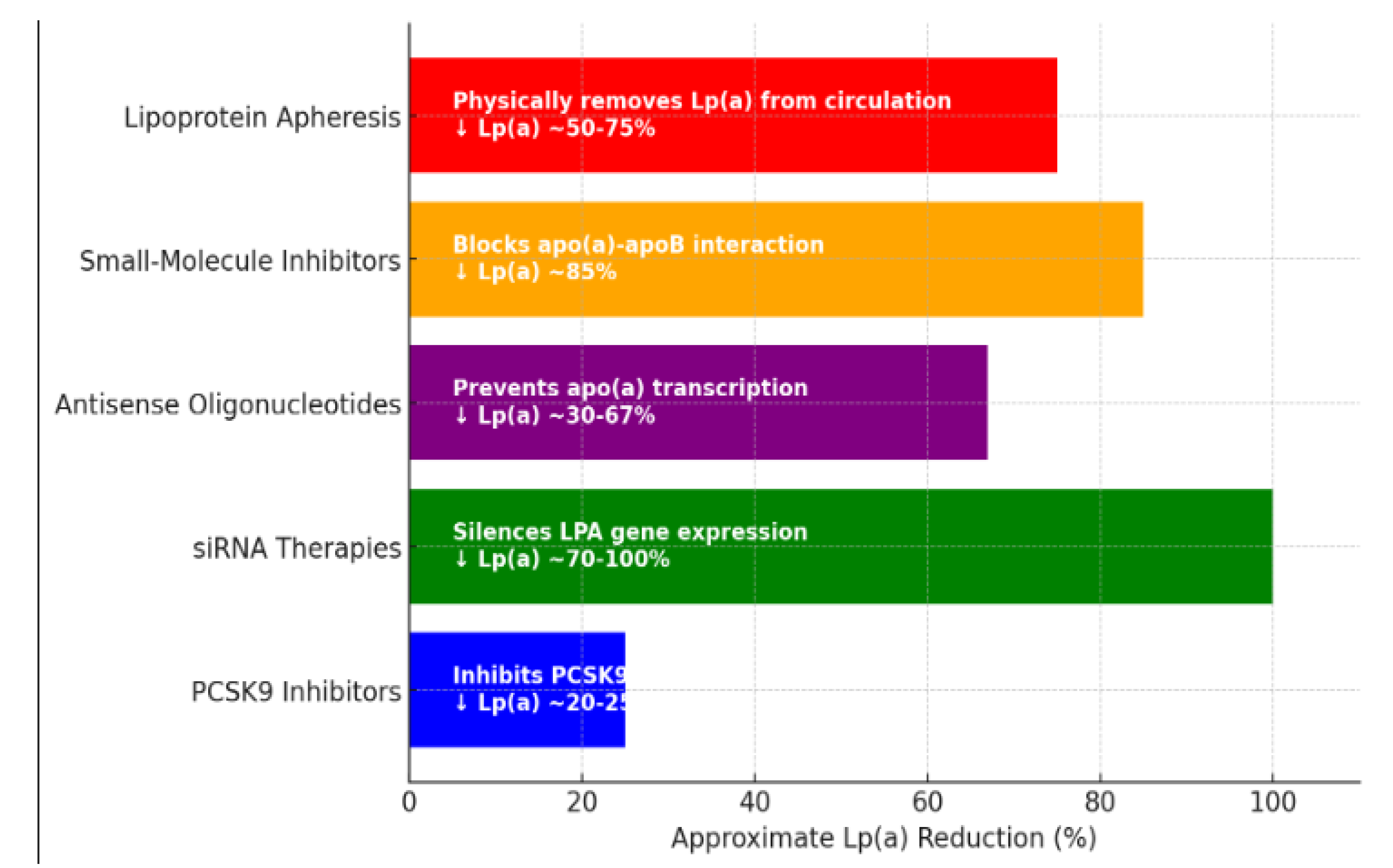
Table 1.
Therapies Targeting Lp(a) Reduction (adapted from reviewed literature).
| Therapy | Mechanism of action | Reduction in Lp(a) | Duration of effect | Limitations |
|---|---|---|---|---|
| PCSK9-i | Increase clearance via LDLR upregulation | 20-25% | short-term, frequent dosing | Moderate effect on Lp(a) reduction |
| ASOs (Pelacarsen) | Inhibits apo(a) mRNA translation | 29–67% | Long-term, monthly dosing | Not yet widely available |
| siRNA (Olpasiran) | Blocks apo(a) mRNA translation | 68.5–100% | Long-term, quarterly dosing | Not yet widely available |
| Apheresis | Physically removes Lp(a) from circulation | up to 73% | Immediate, but transient | Invasive, expensive |
| Small-molecule inhibitors (Muvalaplin) | Disrupts Lp(a) assembly | up to 85.8% | Oral, daily dosing | Early-stage development |
While therapies such as niacin and statins have not demonstrated efficacy in lowering Lp(a), and others, like siRNA, ASO, and muvalaplin, are not yet widely available, PCSK9 inhibitors are accessible treatments with proven Lp(a)-lowering effects. Further studies are required to assess whether this effect could warrant a new indication for this class of medications, independent of LDL cholesterol levels, particularly in high-risk areas, as those in Eastern Europe, allowing patients to bypass the process of trying statins, ezetimibe, and other treatments, which could otherwise delay the benefits [77].
Figure 1.
Therapies Targeting Lp(a) Reduction (adapted from reviewed literature).
4. Conclusion
Lipoprotein(a) has emerged as one of the most promising candidates among biomarkers that could potentially be used for ASCVD screening. The structure of Lp(a) differs from LDL by the presence of apo(a), a protein whose isoform size variability complicates its measurement. Despite the challenges in standardizing Lp(a) assays, significant efforts have been made to improve the accuracy of Lp(a) quantification. Current techniques, such as ELISA, immunoturbidimetry, and liquid chromatography-mass spectrometry (LC-MS/MS), each have their strengths and weaknesses. One limitation that was constant throughout the studies that were reviewed is the variability of apo(a) isoforms, which makes Lp(a) determination difficult.
The clinical relevance of Lp(a) in cardiovascular risk assessment has been intensely studied in recent years. Studies have demonstrated that elevated Lp(a) levels are associated with a higher risk of ASCVD, particularly in individuals with a history of premature or recurrent cardiovascular events. However, despite the growing body of evidence supporting the role of Lp(a) in atherosclerosis, its incorporation into routine clinical practice remains limited. A survey by the European Atherosclerosis Society found that many practitioners still do not routinely measure Lp(a), citing high costs and the absence of targeted treatment options as barriers.
Treatment options for lowering Lp(a) are currently limited. Statins, which are the cornerstone of therapy for dyslipidemia, have minimal impact on Lp(a) levels, with some studies even suggesting a slight increase in Lp(a) with high-intensity statin use. While niacin has shown some potential in reducing Lp(a) levels, the benefits remain controversial and may not translate into significant clinical improvements. Lipoprotein apheresis, a procedure similar to dialysis, has shown promise in reducing both LDL cholesterol and Lp(a), offering cardiovascular benefits such as improved endothelial function and reduced inflammation. However, the complexity and cost of the procedure limit its widespread use.
Recent developments in gene silencing therapies, particularly the use of ASO and siRNA, represent a significant breakthrough in the management of elevated Lp(a). Early-phase clinical trials have shown promising results, with drugs like Pelacarsen demonstrating a 30-80% reduction in Lp(a) levels. While these therapies are still under investigation, they offer a promising alternative for patients with elevated Lp(a) levels who are at high risk of cardiovascular events.
In summary, although Lp(a) is gaining recognition as a significant marker for ASCVD, its routine use in clinical practice remains limited due to challenges related to measurement accuracy and the limited availability of effective treatments. Future research should focus on improving Lp(a) quantification techniques and evaluating the long-term efficacy and safety of gene-silencing therapies. Additionally, further studies are needed to explore the potential of PCSK9 inhibitors to reduce cardiovascular risk in patients independently of LDL levels. With the development of these novel therapies, Lp(a) may soon become a critical target in managing atherosclerotic cardiovascular disease.
Author Contributions
Conceptualization, O.A. and C.G.; methodology, O.A. and G.G.; software, O.A. and O.L.M..; validation, O.A., C.G. and G.G.; formal analysis, C.G. and G.G.; investigation, O.A. and O.L.M.; resources, O.A and O.L.M.; data curation, G.G.; writing—original draft preparation, O.A. and O.L.M; writing—review and editing, O.A. and C.G.; visualization, G.G.; supervision, G.G..; project administration, G.G.; funding acquisition, G.G. All authors performed equal work to the first author. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Nedkoff, L.; Briffa, T.; Zemedikun, D.; Herrington, S.; Wright, F.L. Global Trends in Atherosclerotic Cardiovascular Disease. Clinical Therapeutics 2023, 45, 1087–1091. [Google Scholar] [CrossRef] [PubMed]
- Lyngbakken, M.N.; Myhre, P.L.; Røsjø, H.; Omland, T. Novel Biomarkers of Cardiovascular Disease: Applications in Clinical Practice. Critical Reviews in Clinical Laboratory Sciences 2019, 56, 33–60. [Google Scholar] [CrossRef] [PubMed]
- Geovanini, G.R.; Libby, P. Atherosclerosis and Inflammation: Overview and Updates. Clinical Science 2018, 132, 1243–1252. [Google Scholar] [CrossRef] [PubMed]
- Tasdighi, E.; Adhikari, R.; Almaadawy, O.; Leucker, T.M.; Blaha, M.J. LP(a): Structure, Genetics, Associated Cardiovascular Risk, and Emerging Therapeutics. Annu Rev Pharmacol Toxicol 2024, 64, 135–157. [Google Scholar] [CrossRef]
- Alirocumab and Cardiovascular Outcomes after Acute Coronary Syndrome | New England Journal of Medicine Available online:. Available online: https://www.nejm.org/doi/full/10.1056/NEJMoa1801174 (accessed on 4 March 2025).
- Malick, W.A.; Goonewardena, S.N.; Koenig, W.; Rosenson, R.S. Clinical Trial Design for Lipoprotein(a)-Lowering Therapies: JACC Focus Seminar 2/3. J Am Coll Cardiol 2023, 81, 1633–1645. [Google Scholar] [CrossRef]
- Nissen, S.E.; Wolski, K.; Cho, L.; Nicholls, S.J.; Kastelein, J.; Leitersdorf, E.; Landmesser, U.; Blaha, M.; Lincoff, A.M.; Morishita, R.; et al. Lipoprotein(a) Levels in a Global Population with Established Atherosclerotic Cardiovascular Disease. Open Heart 2022, 9, e002060. [Google Scholar] [CrossRef]
- Catapano, A.L.; Tokgözoğlu, L.; Banach, M.; Gazzotti, M.; Olmastroni, E.; Casula, M.; Ray, K.K. ; Lipid Clinics Network Group Evaluation of Lipoprotein(a) in the Prevention and Management of Atherosclerotic Cardiovascular Disease: A Survey among the Lipid Clinics Network. Atherosclerosis 2023, 370, 5–11. [Google Scholar] [CrossRef]
- Boffa, M.B. Beyond Fibrinolysis: The Confounding Role of Lp(a) in Thrombosis. Atherosclerosis 2022, 349, 72–81. [Google Scholar] [CrossRef]
- Vuorio, A.; Watts, G.F.; Schneider, W.J.; Tsimikas, S.; Kovanen, P.T. Familial Hypercholesterolemia and Elevated Lipoprotein(a): Double Heritable Risk and New Therapeutic Opportunities. J Intern Med 2020, 287, 2–18. [Google Scholar] [CrossRef]
- Duarte Lau, F.; Giugliano, R.P. Lipoprotein(a) and Its Significance in Cardiovascular Disease: A Review. JAMA Cardiology 2022, 7, 760–769. [Google Scholar] [CrossRef]
- Kronenberg, F.; Mora, S.; Stroes, E.S.G.; Ference, B.A.; Arsenault, B.J.; Berglund, L.; Dweck, M.R.; Koschinsky, M.; Lambert, G.; Mach, F.; et al. Lipoprotein(a) in Atherosclerotic Cardiovascular Disease and Aortic Stenosis: A European Atherosclerosis Society Consensus Statement. Eur Heart J 2022, 43, 3925–3946. [Google Scholar] [CrossRef] [PubMed]
- Lipoprotein(a) as a Risk Factor for Cardiovascular Diseases: Pathophysiology and Treatment Perspectives Available online:. Available online: https://www.mdpi.com/1660-4601/20/18/6721 (accessed on 4 March 2025).
- Kronenberg, F. Lipoprotein(a). In Prevention and Treatment of Atherosclerosis: Improving State-of-the-Art Management and Search for Novel Targets; von Eckardstein, A., Binder, C.J., Eds.; Springer: Cham (CH), 2022 ISBN 978-3-030-86075-2.
- Coassin, S.; Kronenberg, F. Lipoprotein(a) beyond the Kringle IV Repeat Polymorphism: The Complexity of Genetic Variation in the LPA Gene. Atherosclerosis 2022, 349, 17–35. [Google Scholar] [CrossRef] [PubMed]
- Enkhmaa, B.; Anuurad, E.; Berglund, L. Lipoprotein (a): Impact by Ethnicity and Environmental and Medical Conditions. J Lipid Res 2016, 57, 1111–1125. [Google Scholar] [CrossRef] [PubMed]
- Awad, K.; Mahmoud, A.K.; Abbas, M.T.; Alsidawi, S.; Ayoub, C.; Arsanjani, R.; Farina, J.M. Intra-Individual Variability in Lipoprotein(a) Levels: Findings from a Large Academic Health System Population. Eur J Prev Cardiol 2024, zwae341. [Google Scholar] [CrossRef]
- Nordestgaard, B.G.; Langsted, A. Lipoprotein(a) and Cardiovascular Disease. The Lancet 2024, 404, 1255–1264. [Google Scholar] [CrossRef]
- Orsó, E.; Schmitz, G. Lipoprotein(a) and Its Role in Inflammation, Atherosclerosis and Malignancies. Clin Res Cardiol Suppl 2017, 12, 31–37. [Google Scholar] [CrossRef]
- Labudovic, D.; Kostovska, I.; Tosheska Trajkovska, K.; Cekovska, S.; Brezovska Kavrakova, J.; Topuzovska, S. Lipoprotein(a) - Link between Atherogenesis and Thrombosis. Prague Med Rep 2019, 120, 39–51. [Google Scholar] [CrossRef]
- Ugovšek, S.; Šebeštjen, M. Lipoprotein(a)—The Crossroads of Atherosclerosis, Atherothrombosis and Inflammation. Biomolecules 2021, 12, 26. [Google Scholar] [CrossRef]
- Enas, E.A.; Varkey, B.; Dharmarajan, T.S.; Pare, G.; Bahl, V.K. Lipoprotein(a): An Independent, Genetic, and Causal Factor for Cardiovascular Disease and Acute Myocardial Infarction. Indian Heart J 2019, 71, 99–112. [Google Scholar] [CrossRef]
- Bu, L.-L.; Yuan, H.-H.; Xie, L.-L.; Guo, M.-H.; Liao, D.-F.; Zheng, X.-L. New Dawn for Atherosclerosis: Vascular Endothelial Cell Senescence and Death. Int J Mol Sci 2023, 24, 15160. [Google Scholar] [CrossRef]
- Emdin, C.A.; Khera, A.V.; Natarajan, P.; Klarin, D.; Won, H.-H.; Peloso, G.M.; Stitziel, N.O.; Nomura, A.; Zekavat, S.M.; Bick, A.G.; et al. Phenotypic Characterization of Genetically Lowered Human Lipoprotein(a) Levels. J Am Coll Cardiol 2016, 68, 2761–2772. [Google Scholar] [CrossRef] [PubMed]
- Kamstrup, P.R.; Nordestgaard, B.G. Elevated Lipoprotein(a) Levels, LPA Risk Genotypes, and Increased Risk of Heart Failure in the General Population. JACC: Heart Failure 2016, 4, 78–87. [Google Scholar] [CrossRef] [PubMed]
- Madsen, C.M.; Kamstrup, P.R.; Langsted, A.; Varbo, A.; Nordestgaard, B.G. Lipoprotein(a)-Lowering by 50 Mg/dL (105 Nmol/L) May Be Needed to Reduce Cardiovascular Disease 20% in Secondary Prevention. Arteriosclerosis, Thrombosis, and Vascular Biology 2020, 40, 255–266. [Google Scholar] [CrossRef] [PubMed]
- Nurmohamed, N.S.; Moriarty, P.M.; Stroes, E.S. Considerations for Routinely Testing for High Lipoprotein(a). Current Opinion in Lipidology 2023, 34, 174. [Google Scholar] [CrossRef]
- Willeit, P.; Ridker, P.M.; Nestel, P.J.; Simes, J.; Tonkin, A.M.; Pedersen, T.R.; Schwartz, G.G.; Olsson, A.G.; Colhoun, H.M.; Kronenberg, F.; et al. Baseline and On-Statin Treatment Lipoprotein(a) Levels for Prediction of Cardiovascular Events: Individual Patient-Data Meta-Analysis of Statin Outcome Trials. Lancet 2018, 392, 1311–1320. [Google Scholar] [CrossRef]
- Minelli, S.; Minelli, P.; Montinari, M.R. Reflections on Atherosclerosis: Lesson from the Past and Future Research Directions. J Multidiscip Healthc 2020, 13, 621–633. [Google Scholar] [CrossRef]
- Li, L.; Liu, S.; Tan, J.; Wei, L.; Wu, D.; Gao, S.; Weng, Y.; Chen, J. Recent Advance in Treatment of Atherosclerosis: Key Targets and Plaque-Positioned Delivery Strategies. J Tissue Eng 2022, 13, 20417314221088509. [Google Scholar] [CrossRef]
- Pirillo, A.; Bonacina, F.; Norata, G.D.; Catapano, A.L. The Interplay of Lipids, Lipoproteins, and Immunity in Atherosclerosis. Curr Atheroscler Rep 2018, 20, 12. [Google Scholar] [CrossRef]
- Medina-Leyte, D.J.; Zepeda-García, O.; Domínguez-Pérez, M.; González-Garrido, A.; Villarreal-Molina, T.; Jacobo-Albavera, L. Endothelial Dysfunction, Inflammation and Coronary Artery Disease: Potential Biomarkers and Promising Therapeutical Approaches. Int J Mol Sci 2021, 22, 3850. [Google Scholar] [CrossRef]
- Ridker, P.M.; MacFadyen, J.G.; Glynn, R.J.; Bradwin, G.; Hasan, A.A.; Rifai, N. Comparison of Interleukin-6, C-Reactive Protein, and Low-Density Lipoprotein Cholesterol as Biomarkers of Residual Risk in Contemporary Practice: Secondary Analyses from the Cardiovascular Inflammation Reduction Trial. Eur Heart J 2020, 41, 2952–2961. [Google Scholar] [CrossRef]
- Buljubasic, N.; Akkerhuis, K.M.; Cheng, J.M.; Oemrawsingh, R.M.; Garcia-Garcia, H.M.; de Boer, S.P.M.; Regar, E.; van Geuns, R.-J.M.; Serruys, P.W.J.C.; Boersma, E.; et al. Fibrinogen in Relation to Degree and Composition of Coronary Plaque on Intravascular Ultrasound in Patients Undergoing Coronary Angiography. Coronary Artery Disease 2017, 28, 23. [Google Scholar] [CrossRef] [PubMed]
- Han, K.; Lu, Q.; Zhu, W.-J.; Wang, T.-Z.; Du, Y.; Bai, L. Correlations of Degree of Coronary Artery Stenosis with Blood Lipid, CRP, Hcy, GGT, SCD36 and Fibrinogen Levels in Elderly Patients with Coronary Heart Disease. Eur Rev Med Pharmacol Sci 2019, 23, 9582–9589. [Google Scholar] [CrossRef] [PubMed]
- Shridas, P.; Tannock, L.R. Role of Serum Amyloid A in Atherosclerosis. Curr Opin Lipidol 2019, 30, 320–325. [Google Scholar] [CrossRef] [PubMed]
- Inflammatory Cytokines and Risk of Coronary Heart Disease: New Prospective Study and Updated Meta-Analysis - PMC Available online:. Available online: https://pmc.ncbi.nlm.nih.gov/articles/PMC3938862/ (accessed on 4 March 2025).
- A Comprehensive 1000 Genomes-Based Genome-Wide Association Meta-Analysis of Coronary Artery Disease - PMC Available online:. Available online: https://pmc.ncbi.nlm.nih.gov/articles/PMC4589895/ (accessed on 4 March 2025).
- Holmes, M.V.; Asselbergs, F.W.; Palmer, T.M.; Drenos, F.; Lanktree, M.B.; Nelson, C.P.; Dale, C.E.; Padmanabhan, S.; Finan, C.; Swerdlow, D.I.; et al. Mendelian Randomization of Blood Lipids for Coronary Heart Disease. Eur Heart J 2015, 36, 539–550. [Google Scholar] [CrossRef]
- Silverman, M.G.; Ference, B.A.; Im, K.; Wiviott, S.D.; Giugliano, R.P.; Grundy, S.M.; Braunwald, E.; Sabatine, M.S. Association Between Lowering LDL-C and Cardiovascular Risk Reduction Among Different Therapeutic Interventions: A Systematic Review and Meta-Analysis. JAMA 2016, 316, 1289–1297. [Google Scholar] [CrossRef]
- Cholesterol Treatment Trialists’ (CTT) Collaboration Efficacy and Safety of More Intensive Lowering of LDL Cholesterol: A Meta-Analysis of Data from 170 000 Participants in 26 Randomised Trials. Lancet 2010, 376, 1670–1681. [CrossRef]
- Marcovina, S.M.; Koschinsky, M.L.; Albers, J.J.; Skarlatos, S. Report of the National Heart, Lung, and Blood Institute Workshop on Lipoprotein(a) and Cardiovascular Disease: Recent Advances and Future Directions. Clinical Chemistry 2003, 49, 1785–1796. [Google Scholar] [CrossRef]
- Marcovina, S.M.; Albers, J.J.; Scanu, A.M.; Kennedy, H.; Giaculli, F.; Berg, K.; Couderc, R.; Dati, F.; Rifai, N.; Sakurabayashi, I.; et al. Use of a Reference Material Proposed by the International Federation of Clinical Chemistry and Laboratory Medicine to Evaluate Analytical Methods for the Determination of Plasma Lipoprotein(a). Clin Chem 2000, 46, 1956–1967. [Google Scholar]
- Lampsas, S.; Xenou, M.; Oikonomou, E.; Pantelidis, P.; Lysandrou, A.; Sarantos, S.; Goliopoulou, A.; Kalogeras, K.; Tsigkou, V.; Kalpis, A.; et al. Lipoprotein(a) in Atherosclerotic Diseases: From Pathophysiology to Diagnosis and Treatment. Molecules 2023, 28, 969. [Google Scholar] [CrossRef]
- Cegla, J.; France, M.; Marcovina, S.M.; Neely, R.D.G. Lp(a): When and How to Measure It. Ann Clin Biochem 2021, 58, 16–21. [Google Scholar] [CrossRef]
- Borque, L.; Rus, A.; del Cura, J.; Maside, C.; Escanero, J. Automated Latex Nephelometric Immunoassay for the Measurement of Serum Lipoprotein (a). J Clin Lab Anal 1993, 7, 105–110. [Google Scholar] [CrossRef] [PubMed]
- Heydari, M.; Rezayi, M.; Ruscica, M.; Jpamialahamdi, T.; Johnston, T.P.; Sahebkar, A. The Ins and Outs of Lipoprotein(a) Assay Methods. Arch Med Sci Atheroscler Dis 2023, 8, e128–e139. [Google Scholar] [CrossRef] [PubMed]
- Szarek, M.; Reijnders, E.; Jukema, J.W.; Bhatt, D.L.; Bittner, V.A.; Diaz, R.; Fazio, S.; Garon, G.; Goodman, S.G.; Harrington, R.A.; et al. Relating Lipoprotein(a) Concentrations to Cardiovascular Event Risk After Acute Coronary Syndrome: A Comparison of 3 Tests. Circulation 2024, 149, 192–203. [Google Scholar] [CrossRef] [PubMed]
- Kamstrup, P.R. Lipoprotein(a) and Cardiovascular Disease. Clinical Chemistry 2021, 67, 154–166. [Google Scholar] [CrossRef]
- Shahjehan, R.D.; Sharma, S.; Bhutta, B.S. Coronary Artery Disease. In StatPearls; StatPearls Publishing: Treasure Island (FL), 2025. [Google Scholar]
- O’Donoghue, M.L.; Rosenson, R.S.; Gencer, B.; López, J.A.G.; Lepor, N.E.; Baum, S.J.; Stout, E.; Gaudet, D.; Knusel, B.; Kuder, J.F.; et al. Small Interfering RNA to Reduce Lipoprotein(a) in Cardiovascular Disease. New England Journal of Medicine 2022, 387, 1855–1864. [Google Scholar] [CrossRef]
- Chan, D.C.; Watts, G.F. The Promise of PCSK9 and Lipoprotein(a) as Targets for Gene Silencing Therapies. Clin Ther 2023, 45, 1034–1046. [Google Scholar] [CrossRef]
- 2019 ESC/EAS Guidelines for the Management of Dyslipidaemias: Lipid Modification to Reduce Cardiovascular Risk | European Heart Journal | Oxford Academic Available online:. Available online: https://academic.oup.com/eurheartj/article/41/1/111/5556353 (accessed on 4 March 2025).
- Lampsas, S.; Xenou, M.; Oikonomou, E.; Pantelidis, P.; Lysandrou, A.; Sarantos, S.; Goliopoulou, A.; Kalogeras, K.; Tsigkou, V.; Kalpis, A.; et al. Lipoprotein(a) in Atherosclerotic Diseases: From Pathophysiology to Diagnosis and Treatment. Molecules 2023, 28, 969. [Google Scholar] [CrossRef]
- A, S.; Ž, R.; Le, S.-M.; G, F.; Af, C. Effect of Extended-Release Niacin on Plasma Lipoprotein(a) Levels: A Systematic Review and Meta-Analysis of Randomized Placebo-Controlled Trials. Metabolism: clinical and experimental 2016, 65. [Google Scholar] [CrossRef]
- Schwartz, G.G.; Ballantyne, C.M. Existing and Emerging Strategies to Lower Lipoprotein(a). Atherosclerosis 2022, 349, 110–122. [Google Scholar] [CrossRef]
- Tsaban, G. Statins and Lipoprotein(a); Facing the Residual Risk. European Journal of Preventive Cardiology 2022, 29, 777–778. [Google Scholar] [CrossRef]
- de Boer, L.M.; Oorthuys, A.O.J.; Wiegman, A.; Langendam, M.W.; Kroon, J.; Spijker, R.; Zwinderman, A.H.; Hutten, B.A. Statin Therapy and Lipoprotein(a) Levels: A Systematic Review and Meta-Analysis. European Journal of Preventive Cardiology 2022, 29, 779–792. [Google Scholar] [CrossRef] [PubMed]
- Safarova, M.S.; Moriarty, P.M. Lipoprotein Apheresis: Current Recommendations for Treating Familial Hypercholesterolemia and Elevated Lipoprotein(a). Curr Atheroscler Rep 2023, 25, 391–404. [Google Scholar] [CrossRef] [PubMed]
- Víšek, J.; Bláha, M.; Bláha, V.; Lášticová, M.; Lánska, M.; Andrýs, C.; Tebbens, J.D.; Igreja e Sá, I.C.; Tripská, K.; Vicen, M.; et al. Monitoring of up to 15 Years Effects of Lipoprotein Apheresis on Lipids, Biomarkers of Inflammation, and Soluble Endoglin in Familial Hypercholesterolemia Patients. Orphanet J Rare Dis 2021, 16, 110. [Google Scholar] [CrossRef]
- Padmanabhan, A.; Connelly-Smith, L.; Aqui, N.; Balogun, R.A.; Klingel, R.; Meyer, E.; Pham, H.P.; Schneiderman, J.; Witt, V.; Wu, Y.; et al. Guidelines on the Use of Therapeutic Apheresis in Clinical Practice - Evidence-Based Approach from the Writing Committee of the American Society for Apheresis: The Eighth Special Issue. J Clin Apher 2019, 34, 171–354. [Google Scholar] [CrossRef] [PubMed]
- Thompson, G.R. The Scientific Basis and Future of Lipoprotein Apheresis. Ther Apher Dial 2022, 26, 32–36. [Google Scholar] [CrossRef]
- Nugent, A.K.; Gray, J.V.; Gorby, L.K.; Moriarty, P.M. Lipoprotein Apheresis: First FDA Indicated Treatment for Elevated Lipoprotein(a). J Clin Cardiol 2020, Volume 1, 16–21. [Google Scholar] [CrossRef]
- Zimmermann, T.S.; Karsten, V.; Chan, A.; Chiesa, J.; Boyce, M.; Bettencourt, B.R.; Hutabarat, R.; Nochur, S.; Vaishnaw, A.; Gollob, J. Clinical Proof of Concept for a Novel Hepatocyte-Targeting GalNAc-siRNA Conjugate. Mol Ther 2017, 25, 71–78. [Google Scholar] [CrossRef]
- Macchi, C.; Sirtori, C.R.; Corsini, A.; Santos, R.D.; Watts, G.F.; Ruscica, M. A New Dawn for Managing Dyslipidemias: The Era of Rna-Based Therapies. Pharmacol Res 2019, 150, 104413. [Google Scholar] [CrossRef]
- Landmesser, U.; Poller, W.; Tsimikas, S.; Most, P.; Paneni, F.; Lüscher, T.F. From Traditional Pharmacological towards Nucleic Acid-Based Therapies for Cardiovascular Diseases. European Heart Journal 2020, 41, 3884–3899. [Google Scholar] [CrossRef]
- Katzmann, J.L.; Packard, C.J.; Chapman, M.J.; Katzmann, I.; Laufs, U. Targeting RNA With Antisense Oligonucleotides and Small Interfering RNA: JACC State-of-the-Art Review. J Am Coll Cardiol 2020, 76, 563–579. [Google Scholar] [CrossRef]
- Yeang, C.; Karwatowska-Prokopczuk, E.; Su, F.; Dinh, B.; Xia, S.; Witztum, J.L.; Tsimikas, S. Effect of Pelacarsen on Lipoprotein(a) Cholesterol and Corrected Low-Density Lipoprotein Cholesterol. J Am Coll Cardiol 2022, 79, 1035–1046. [Google Scholar] [CrossRef] [PubMed]
- Novartis Pharmaceuticals A Randomized Double-Blind, Placebo-Controlled, Multicenter Trial Assessing the Impact of Lipoprotein (a) Lowering With Pelacarsen (TQJ230) on Major Cardiovascular Events in Patients With Established Cardiovascular Disease; clinicaltrials. 2025.
- Katsiki, N.; Vrablik, M.; Banach, M.; Gouni-Berthold, I. Inclisiran, Low-Density Lipoprotein Cholesterol and Lipoprotein (a). Pharmaceuticals (Basel) 2023, 16, 577. [Google Scholar] [CrossRef]
- Sabatine, M.S.; Giugliano, R.P.; Keech, A.C.; Honarpour, N.; Wiviott, S.D.; Murphy, S.A.; Kuder, J.F.; Wang, H.; Liu, T.; Wasserman, S.M.; et al. Evolocumab and Clinical Outcomes in Patients with Cardiovascular Disease. New England Journal of Medicine 2017, 376, 1713–1722. [Google Scholar] [CrossRef] [PubMed]
- O’Donoghue, M.L.; Rosenson, R.S.; Gencer, B.; López, J.A.G.; Lepor, N.E.; Baum, S.J.; Stout, E.; Gaudet, D.; Knusel, B.; Kuder, J.F.; et al. Small Interfering RNA to Reduce Lipoprotein(a) in Cardiovascular Disease. N Engl J Med 2022, 387, 1855–1864. [Google Scholar] [CrossRef]
- Eli Lilly and Company A Phase 2, Randomized, Double-Blind, Placebo-Controlled Study to Investigate the Efficacy and Safety of LY3819469 in Adults With Elevated Lipoprotein(a); clinicaltrials. 2025.
- Nissen, S.E.; Linnebjerg, H.; Shen, X.; Wolski, K.; Ma, X.; Lim, S.; Michael, L.F.; Ruotolo, G.; Gribble, G.; Navar, A.M.; et al. Lepodisiran, an Extended-Duration Short Interfering RNA Targeting Lipoprotein(a). JAMA 2023, 330, 2075–2083. [Google Scholar] [CrossRef]
- Hooper, A.J.; Fernando, P.M.S.; Burnett, J.R. Potential of Muvalaplin as a Lipoprotein(a) Inhibitor. Expert Opinion on Investigational Drugs 2024, 33, 5–7. [Google Scholar] [CrossRef]
- Nicholls, S.J.; Ni, W.; Rhodes, G.M.; Nissen, S.E.; Navar, A.M.; Michael, L.F.; Haupt, A.; Krege, J.H. Oral Muvalaplin for Lowering of Lipoprotein(a): A Randomized Clinical Trial. JAMA 2025, 333, 222–231. [Google Scholar] [CrossRef]
- SCORE2-OP Risk Prediction Algorithms: Estimating Incident Cardiovascular Event Risk in Older Persons in Four Geographical Risk Regions. Eur Heart J 2021, 42, 2455–2467. [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



