Submitted:
08 March 2024
Posted:
11 March 2024
You are already at the latest version
Abstract
Cerebellar ataxia, neuronopathy, and vestibular areflexia syndrome (CANVAS) is caused by biallelic pathogenic expansions, or compound heterozygosity with other pathogenic variants in the RFC1 gene. CANVAS is estimated to be underdiagnosed, both because of the lack of formal diagnostic criteria and molecular challenges that translate to lesser access and high cost of routine testing. Our aim was to address the need for making CANVAS genetic testing routine, by designing a streamlined two-step PCR consisting of a short-allele screening PCR and a confirmatory PCR with fragment capillary electrophoresis detection. Exome sequencing of RFC1 was additionally foreseen to resolve potential compound heterozygosity cases. Specificity of our approach was evaluated using ataxia patients with known non-CANVAS diagnoses, and optimized using Southern blot confirmed CANVAS patients. We evaluated our approach by testing patients consecutively referred for clinically suspected CANVAS using first the two-step PCR, followed by exome sequencing. Our approach was able to accurately identify negative and confirm positive cases in prospectively collected suspected CANVAS patients presenting with at least three typical clinical signs. The proposed testing approach provides an alternative method able to clearly distinguish between CANVAS negative and positive cases and can be easily incorporated into the genetic diagnostic laboratory workflow.
Keywords:
Cerebellar ataxia neuronopathy and vestibular areflexia syndrome (CANVAS)
; RFC1
; genetic testing
; genetic diagnostics
; fluorescent PCR
; repeat-primed PCR
; repeat-expansion
; fragment length analysis
Introduction
The initial delineation of a distinct syndrome encompassing cerebellar and bilateral vestibular dysfunction was first reported in 2004 (Migliaccio et al., 2004). In 2011, the name cerebellar ataxia, neuropathy, and vestibular areflexia syndrome (acronym CANVAS) was proposed (Szmulewicz et al., 2011), and the causative biallelic, intronic, pathogenic repeat expansion sequence in the replication factor C subunit 1 (RFC1) gene was finally identified in 2019 (Cortese et al., 2019). Subsequently, clinical presentation associated with biallelic RFC1 (AAGGG)n repeat expansions was broadened to include sensory, cerebellar, and vestibular dysfunction, and impairment of only one of the balance control systems. Chronic cough and autonomic dysfunction were also occasionally described (Wu et al., 2014). Further supportive findings from diagnostic workup mainly include reduced or absent sensory action potential, cerebellar atrophy, and vestibular testing showing bilateral vestibular dysfunction (Szmulewicz et al., 2011; Cortese et al., 2019, 2020). A recessive mode of inheritance is expected, whereas the absence of a known family history does not exclude the diagnosis. Official guidelines for diagnostic criteria for CANVAS syndrome still need to be established.
Current estimation of the CANVAS syndrome prevalence ranges from 1:10 000 to 1:625 individuals (Cortese et al., 2020, 2023), which indicates that this is an underdiagnosed disorder, likely due to its clinical and genetic heterogeneity posing a diagnostic challenge. Until recently, the diagnosis of CANVAS syndrome was based solely on clinical presentation. While the identification of causative biallelic (AAGGG)n repeat expansions in the RFC1 gene enabled genetic confirmation of suspected CANVAS cases, it also introduced new challenges, clearly suggesting a much broader phenotypic spectrum of associated disorders (Cortese et al., 2019, 2020). Recent studies showed that only 23% of patients with genetically confirmed CANVAS were previously clinically suspected to have CANVAS (Cortese et al., 2020). Moreover, only two-thirds of cases presented with complete CANVAS phenotype, whereas others presented with sensory neuropathy, either as a sole manifestation or in combination with cerebellar or vestibular impairment. Interestingly, several recent case and series reports suggest RFC1 pathogenic expansions may be present in patients with other disorders. (Kytövuori et al., 2022; Fonte et al., 2023; Hirano et al., 2023). All this highlights the importance of genetic testing for all cases with suggestive findings that could enable us to detect positive CANVAS patients and exclude patients who do not have CANVAS (Hadjivassiliou et al., 2023).
In addition to the highly variable phenotypic spectrum, there are several molecular challenges to the routine genetic diagnostics of CANVAS. These include the relatively high prevalence of non-pathogenic expansions in the RFC1 gene, as well as the sequence heterogeneity of non-pathogenic expansions, interruptions, mixed repeat motifs of the sequence expansions, and their varying size (Cortese et al., 2023). Additionally, recently single nucleotide frameshift and truncating variants have been shown as pathogenic (Ronco et al., 2023), and despite being rare, they also now need to be considered in diagnostic testing.
The original methodology developed in 2019, and since used for research purposes (Akçimen et al., 2019; Cortese et al., 2019, 2023; Dominik et al., 2023), has not been designed with the exclusive aim of routine diagnostic exclusion or confirmation of CANVAS and so consists of a combinatory approach of flanking PCR (detected on the gel where similar length short alleles are not distinguishable), RP-PCR targeting one of the three possible nucleotide sequences (anchor primer system with specificity issues), as well as Southern blot (for determining length) and Sanger sequencing (for additional confirmation).
The limitations of the original PCR which uses gel detection of short variants and an RT-PCR anchor method with multiple, partially overlapping primers per type of sequence repeat, have been well described (Cortese et al., 2019), limiting the use of this method in expansive testing of patients with limited phenotypes in particular and in diagnostic testing where other disorders presenting with ataxia, such as Friedreich’s ataxia or various spinocerebellar ataxias. These disease, that are also caused by different expansions with what may be similar sequences, may present an additional diagnostic challenge.
While some novel methods, such as short-read whole genome sequencing (Bergant et al., 2021) and optical genome mapping (Ghorbani et al., 2022), can be used to either detect or size the expanded repeats, neither method at the moment is confirmatory by itself. Others, like long-read sequencing (Nakamura et al., 2020; Dominik et al., 2023), can both size and determine the sequence of the repeat and have been used successfully to do so; however, at the moment, their cost is prohibitive to routine use.
Our aim was to address the lack of accessibility of routine CANVAS genetic testing, by designing a streamlined two-step PCR consisting of a short-allele screening PCR and a confirmatory PCR with fragment capillary electrophoresis detection. The test was designed with the aim of clearly distinguishing CANVAS negative from CANVAS positive cases in a routine setting. The test was optimized using Southern blot confirmed CANVAS cases. The specificity of the test was evaluated using 19 ataxia cases with known non-CANVAS genetic causes. To evaluate our improved diagnostic approach, we have used our PCR approach to test 15 new consecutively referred patients with clinically suspected CANVAS, with RFC1 sequencing follow-up in heterozygote carriers to test for pathogenic single nucleotide variants.
Results
Testing Design Rationale and Interpretation
The design of the CANVAS testing approach is presented in Figure 1. The testing design rationale is such that in the first-step PCR, all individuals with alleles below the proposed pathogenic length of 250 repeats can be identified. The resolution of the fragment length analysis enables distinction between alleles differing in size by one repeat length, which is an improvement from the original method, that is based on gel detection. Both of the primers are labelled to ensure amplicon specificity from both ends, which is preferable given the high variability of the region. In this way, when two shorter PCR products are detected using both labels, biallelic CANVAS RFC1 intronic expansion can be conclusively excluded.
The rationale of the second step or confirmatory PCR is such that individuals with only one short allele (below 250 bp) or no short alleles detected in the first step (expansion candidates) are tested for the presence of pathogenic expansion sequence. Because of the high prevalence of non-pathogenic repeat sequences (AAAAG)n, the testing for the three most common expansion sequences, (AAAAG)n, (AAGGG)n, and (AAAGG)n is performed in parallel. In the second step RT-PCRs, the same specific labeled reverse primer used in the initial PCR or screening reaction is used, ensuring that only true positive expanded sequences at the correct RFC1 location are detected. In this way, false positives due to repeat sequences located elsewhere cannot be detected using fragment length analysis, as they do not produce a fluorescent PCR product. The full two-step PCR principle is presented in Figure 2.
The interpretation of PCR results is also made in two steps (Figure 1). First, the presence of short, normal alleles in homozygous or heterozygous form is confirmed (negative CANVAS or candidate CANVAS), or the presence of short alleles is entirely excluded (candidate CANVAS). Next, in the second PCR step, three expansion PCRs, sequence-specific for the three most common repeat motifs (AAAAG/AAAGG/AAGGG)n are performed, enabling sequence-specific confirmation of the repeat present. At this moment, the biallelic (AAGGG)n expansion is considered pathogenic or CANVAS positive, while the (AAAGG)n expansion is considered pathogenic depending on its size (Dominik et al., 2023). The other alternative pathogenic repeats that have been proposed occur rarely, but can be incorporated easily into our second-step PCR through additional unlabeled primers should the need occur.
Finally, as a third step, in order to address possible compound heterozygosity with frameshift and terminating RFC1 variants in trans, in patients who are carriers/heterozygotes for the pathogenic sequence expansion, exome sequencing with initial analysis of the RFC1 gene should be performed, to finally confirm or exclude CANVAS. In case of negative exome sequencing result for RFC1 pathogenic variants, the testing can be easily expanded to other genes, as is the current standard approach in genetic testing of ataxia in general, as proposed by the European Reference Network for Rare Neurological Diseases (ERN-RN) (https://www.ern-rnd.eu/wp-content/uploads/2023/07/Final-Flowchart-adult-Ataxias_en.pdf.
Specificity of Testing Results
A total of 19 ataxia patients with known non-CANVAS diagnoses were tested to evaluate the sensitivity of the two-step approach, and all could be excluded as having CANVAS (Table 1). Our test was able to successfully exclude CANVAS in these patients, despite a high proportion of non-pathogenic expansions in this non-CANVAS cohort. Of the tested non-CANVAS ataxia patients, 47.4 % (9/19) had monoallelic non-pathogenic (AAAAG)n expansions, and 10.5 % (2/19) had a biallelic non-pathogenic (AAAAG)n expansion.
Prospective Testing Results
Among the 15 ataxia patients consecutively referred for testing because of clinical suspicion of CANVAS, 5 (33.3 %) were confirmed as having biallelic pathogenic (AAGGG)n expansions (Table 2), while one patient had mixed sequence expansions including the non-pathogenic repeat, and one patient did not have any of the tested three expansion sequences. While this patient did not provide consent for exome sequencing, the patient heterozygous for the (AAAGG)n expansion was additionally tested using a gene panel exome sequencing containing the RFC1 gene, and heterozygosity with single nucleotide variants was excluded, so far excluding a genetic diagnosis of CANVAS.
Currently, such atypical expansion profiles are not considered to constitute the genetic cause of CANVAS, however, their possible partial contribution to this complex phenotype may be determined in further studies. Similar to the CANVAS negative cohort, the (AAAAG)n expansions were common, and were found to be present in 40.0% (6/15) of the tested patients.
The clinical characteristics of the five CANVAS-positive patients identified by our two-step approach are given in Table 3.
A First-step PCR principle detects short alleles with high specificity and sensitivity, resulting from the simultaneous detection of dual-labeled peaks. B Second step PCRs can distinguish the three most common expansion motifs. C Example results of first-step PCR showing the detection of short and expanded alleles. Expanded alleles in the technical context are alleles above the reference n =11 size, and can be detected up to n=250. The absence of any PCR products in the first-step PCR indicates a high likelihood of biallelic expansion. D Example of second step expansion PCRs of an individual without short alleles, showing (AAAAG)n and (AAAGG)n expansion in the absence of pathogenic (AAGGG)n repeats. E Example of second step expansion PCRs of an individual without short alleles, showing the pathogenic (AAGGG)n expansion. Complete lack of short PCR amplification with the same labelled primer indicates the expansion is biallelic.
Discussion
Currently, conventional PCR, repeat-primed PCR, and Southern blotting (Akçimen et al., 2019; Cortese et al., 2019, 2023) are used to detect pathogenic repeats, size them and confirm biallelic composition. However, confirmation of biallelic pathogenic expansion is difficult in a routine diagnostic setting where many referral diagnoses may overlap or clinical data may not be readily available to the testing laboratory. While several novel methods, such as whole genome sequencing (Bergant et al., 2021), and optical genome mapping (Ghorbani et al., 2022), can be used to either detect or size the expanded repeats, and long-read sequencing can do both (Nakamura et al., 2020; Dominik et al., 2023), their cost at the moment remains prohibitive to expanding screening and their incorporation into routine diagnostics.
In routine diagnostics, the purpose of the testing is to identify negative and positive patients according to the current knowledge of the genetics of the disease. While this was the primary aim of our diagnostic design, we would like to point out that in expansion disorders in particular, this goal often represents a moving target, with constant novel insights (Dominik et al., 2023; Ronco et al., 2023; Currò et al., 2024).
While the original pathogenic (AAGGG)n expansions in the RFC1 gene, most commonly ranging from 400 to more than 2000 repeats, were reported in 82%-97% of cases with typical CANVAS phenotype (Cortese et al., 2019; Rafehi et al., 2019), subsequent studies have identified many rare additional possibly causative RFC1 repeat configurations (Cortese et al., 2019; Beecroft et al., 2020; Scriba et al., 2020; Tsuchiya et al., 2020; Dominik et al., 2023). These include (ACAGG)n, (AGGGC)n, as well as interrupted repeats, such as (AAAGG)10-25(AAGGG)exp (AAAGG)4-6, and some rare sequence variants, such as (AGAGG)n, (AAGGC)n so far only observed in single families. Recently, the previously non-pathogenic (AAAGG)n expansions (ranging from 40 to 1000 repeats), have now been shown to have a size-dependent pathogenicity (Dominik et al., 2023), which likely depends on their ability to form G-quadruplex structures (Dominik et al., 2023; Currò et al., 2024).
Furthermore, the recent discovery that compound heterozygosity for pathogenic RFC1 repeats and truncating variants on the other RFC1 allele also result in CANVAS has expanded our understanding of its genetic landscape (Ronco et al., 2023). Therefore, it is essential to note that even presumed heterozygote carriers of the pathogenic expansion allele need to be tested for additional single nucleotide pathogenic variants on the other allele. In order to account for this possibility, we propose all heterozygous carriers of the pathogenic repeat expansion should be tested for single nucleotide variants on the other allele, in order to exclude compound heterozygosity (Ronco et al., 2023). This is also something that has been suggested by the previous Reviewers. The reason we have chosen exome sequencing over target sequencing of the gene is that exome sequencing is now one of the standard approaches in genetic testing of ataxia in general as put forward by the European Reference Network for Rare Neurological Diseases (ERN-RN) (https://www.ern-rnd.eu/wp-content/uploads/2023/07/Final-Flowchart-adult-Ataxias_en.pdf. In cases where CANVAS is not confirmed, this approach enables us to look for other causes of ataxia in our patients by expanding the interpretation to additional gene panels.
Designed with the routine diagnostic aim in mind, our two-step PCR approach was able, in the highly specific dual fluorescently labeled first-step, to accurately identify as negative all non-CANVAS cases with previously known genetic causes, including those with pathogenic expansions in other genes (e.g. Friedreich ataxia, spinocerebellar ataxias) (Table 1). In line with previous studies, we also observed a high prevalence of expansions using the first-step PCR, which was shown as sequence-wise non-pathogenic (AAAAG)n expansions in the second-step PCR, while the presence of the pathogenic (AAGGG)n expansions could be excluded with confidence.
On the other hand, in the prospectively collected cases with clinical suspicion of CANVAS, we were able, by using our second step PCR, optimized on Southern blot confirmed cases, to distinguish the three most common repeat sequences from each other, and to accurately identify cases with clear pathogenic biallelic expansions. In this way, we have confirmed the presence of biallelic (AAGGG)n pathogenic repeats in five out of 15 cases, while identifying the allelic composition in all but one case, that likely has an atypical and yet unknown expansion sequence, which we plan to investigate further. While our method has the potential to detect heterozygote carriers with one typical pathogenic expansion allele, in the presence of normal or non-pathogenic expansion on the other allele, we have only observed this situation in one of our limited number of cases. In the one patient, we observed to be heterozygous for the repeat-size dependent pathogenic (AAAGG)n repeat, exome sequencing did not identify any pathogenic SNV variants in RFC1.
The limitation of our approach is the detection of the three most common sequence repeats. Notably, other repeat sequences, and mixed sequence repeat profiles at this location have already been observed in different populations (Cortese et al., 2023), however known pathogenic sequences so far remain rare, and mostly limited geographically or to single families (Dominik et al., 2023). While our PCR setup doesn’t allow us to diagnostically categorize such rare patients, it enables them to be included in further research, or to be flagged for testing with additional methods. Furthermore, despite the stated limitations, the design of our second-step PCR is such, that any additional sequences identified as pathogenic in the future, can easily be added in the way of an unlabeled forward primer, should they be identified as important in our population. Furthermore, it needs to be emphasized that unless shown to affect RFC1 by functional analysis as has been performed for some of these rare variants by other groups (Dominik et al., 2023), patients with novel expansions lacking any of the known pathogenic repeats should be considered as CANVAS negative, until proof of the association with the complex presenting phenotypes, which is not the primary aim of diagnostic testing in this underreported disorder. In all cases, causality must be established for patients that may not fit into clearly negative or positive categories by additional confirmation. On the other hand, to the best of our knowledge, no healthy individuals with a (AAGGG)n pathogenic expansion, lacking a shorter allele or a non-pathogenic expansion on the other allele (i.e. having an unknown expansion on the other allele), have yet been identified. Therefore, if there are any heterozygotes of (AAGGG)n expansion and possible unknown expansion with a clinical CANVAS presentation, they would currently be considered as CANVAS positive.
Indeed, the importance of clinical symptoms must be highlighted, since clinical presentation described in patients with genetically confirmed CANVAS was suggestive. Namely, all of them presented with at least three of the typical symptoms, including cerebellar ataxia, bilateral vestibular areflexia, sensory neuropathy, autonomic dysfunction, and/or chronic cough. On the other hand, patients negative for CANVAS all presented with only one or two typical symptoms, whereas only one presented with autonomic dysfunction, and none were referred with symptoms of vestibular dysfunction (results of vestibular testing were either negative or missing). Still, as previously discussed, the observed clinical presentation may place many patients in a broader CANVAS phenotype specter and thus genetic testing is necessary to finally exclude/confirm CANVAS diagnosis, which is what our diagnostic setup now enables.
Conclusion
The streamlined two-step fragment analysis PCR supplemented with exome sequencing represents a fast and accurate method for routine genetic diagnostic testing in suspected CANVAS patients, that clearly distinguishes negative and positive patients, as well as those patients where further research investigation is needed. We propose that suspected CANVAS patients with heterozygous pathogenic expansions be tested with exome sequencing for identification or exclusion of possible compound heterozygosity with pathogenic single-nucleotide variants.
Materials and Methods
Patient Cohort
The material of all individuals tested in the study was collected as part of their routine genetic testing and written informed consents for genetic testing/and or exome sequencing and the use of the samples in research and design of diagnostic tests was obtained from the patients during their clinical appointment, granting the rights to publish the findings of genetic testing in de-identified form in scientific literature. The informed consent statements were prepared according to the National Review Board guidelines and approved by the Institutional Ethics Board at the University Medical Centre Ljubljana, Slovenia. All medical procedures in the study were performed according to the ethical standards of the Helsinki Declaration and all applying national regulations.
Three patients with Southern blot-confirmed CANVAS were used as positive controls and to optimize the PCR conditions. A total of 19 ataxia patients with confirmed non-CANVAS diagnosis (Friedreich Ataxia, Spinocerebellar ataxias, etc.) were used to confirm the specificity of the test for CANVAS. Finally, 15 patients consecutively referred to the CIGM due to clinically suspected CANVAS between Jan 2021 and Jan 2023 were tested using the two-step PCR approach and exome sequencing when required.
DNA Extraction
Genomic DNA was extracted from peripheral blood using QuickGene DNA whole blood kit L (DB-L) on Kurabo Biomedical QuickGene-610L Nucleic Acid Isolation System (Kurabo, Japan) and quantified by the Qubit® 2.0 Fluorometer (Thermo Fisher Scientific, USA) according to the manufacturer’s instructions.
Primer Design
The primers used in the study were designed manually onto the least variable regions in the RFC1 adjacent to the location of the RFC1 intronic repeats. The melting temperature and absence of secondary structures using NetPrimer (Premier Biosoft International) and TIB-Molbiol (Berlin, Germany) free web-based software. The ‘Primer Pair Specificity Checking Parameters’ function of Primer-BLAST (NCBI, https://www.ncbi.nlm.nih.gov/) was used to extensively check for the specificity of the primers. The primer pairs used for the first-step short PCR (PCR1) and confirmatory fragment analysis PCRs (PCR 2-4) are given in Table 4.
Two-Step PCR Design and Conditions
The two-step PCR approach consists of the first-step to detect short alleles (non-expanded) and, by this, to exclude CANVAS RFC1 intronic expansion, and the second step or confirmatory PCR to determine the presence of non-pathogenic and pathogenic expansion sequences (AAAAG/AAGGG/AAAGG) in patients missing short PCR products. The fluorescent labeling of both primers in the first-step PCR ensures the specificity of the reaction, while in the next step, only the reverse primer, which is the same as in the screening reaction, remains labeled so that no false positives due to repeat sequences located elsewhere are detected.
The short-PCR was carried out in a 10 µl reaction volume containing 50 ng genomic DNA, 0.02 U/µl KOD Xtreme™ Hot Start DNA Polymerase (Sigma-Aldrich), 1x Xtreme™ Buffer, 0.4 µM of dNTPs (each), and 0.1 µM of RFC1-FW-FAM 5’-FAM- CTGAGGCAGGAGATTCACTTG-3’ primer and 0.2 µM of RFC1-REV-VIC 5’-VIC-GGTGGCTGTCTCATCTGTTG-3’ primer.
The thermal cycler program was set to 2 min at 94°C, followed by 5 cycles consisting of 10s at 98°C, 30 s at 68°C ramp to 63°C, and 40 s at 68°C, followed by 35 cycles consisting of 10s at 98°C, 30 s at 63°C, and 40 s at 68°C. The final extension step was performed at 68°C for 10 min, and the reaction mixtures were then cooled to 8°C.
PCR products were resolved by capillary electrophoresis using the Applied Biosystems® 3500 Genetic Analyzer according to the manufacturer's instructions. In the case of two different FAM and VIC labeled products, confirmatory PCRs for AAAAG/AAAGG/AAGGG expansions were carried out in parallel in the following way. Each repeat sequence expansion was amplified by long-range PCR, using the optimized protocol for a 10 µl reaction volume containing 50 ng genomic DNA, 0.02 U/µl KOD Xtreme™ Hot Start DNA Polymerase (Sigma-Aldrich), 1x Xtreme™ Buffer, 0.4 µM of dNTPs (each), and 0.1 µM of appropriate primer 5’-RFC1-RP-AAAAG-F GAAAAGAAAAGAAAAGAAAAGAAAAG-3’ or 5’-RFC1-RP-AAAGG-F AAAGGAAAGGAAAGGAAAGG-3’ or 5’-RFC1-RP-AAGGG-F GAAGGGAAGGGAAGGGAA-3’ and 0.2 µM of the reverse 5’-RFC1-REV-VIC GGTGGCTGTCTCATCTGTTG-3’ primer, in each of the PCRs.
The thermal cycler program was set to 2 min at 94°C, followed by 5 cycles consisting of 10s at 98°C, 30 s at 68°C ramp to 63°C, and 40 s at 68°C, followed by 35 cycles consisting of 10s at 98°C, 30 s at 63°C, and 40 s at 68°C. The final extension step was performed at 68°C for 10 min, and the reaction mixtures were then cooled to 8°C.
Per the manufacturer's instructions, PCR products were resolved by capillary electrophoresis using the Applied Biosystems® 3500 Genetic Analyzer.
Exome Sequencing
Short-read Illumina exome sequencing was performed at the Clinical Insitute of Genomic Medicine, University Medical Centre Ljubljana, Slovenia (CIGM, UMCL), as previously described (Babić Božović et al., 2021). Variants in the RFC1 gene were classified according to the ACMG and AMP 2015 joint consensus recommendation (The ACMG Laboratory Quality Assurance Committee et al., 2015).
References
- Akçimen, F., Ross, J. P., Bourassa, C. V., Liao, C., Rochefort, D., Gama, M. T. D., et al. (2019). Investigation of the RFC1 Repeat Expansion in a Canadian and a Brazilian Ataxia Cohort: Identification of Novel Conformations. Front Genet 10, 1219. [CrossRef]
- Babić Božović, I., Maver, A., Leonardis, L., Meznaric, M., Osredkar, D., and Peterlin, B. (2021). Diagnostic yield of exome sequencing in myopathies: Experience of a Slovenian tertiary centre. PLoS One 16, e0252953. 5295. [CrossRef]
- Beecroft, S. J., Cortese, A., Sullivan, R., Yau, W. Y., Dyer, Z., Wu, T. Y., et al. (2020). A Māori specific RFC1 pathogenic repeat configuration in CANVAS, likely due to a founder allele. Brain 143, 2673–2680. [CrossRef]
- Bergant, G., Maver, A., and Peterlin, B. (2021). Whole-Genome Sequencing in Diagnostics of Selected Slovenian Undiagnosed Patients with Rare Disorders. Life 11, 205. [CrossRef]
- Cortese, A., Reilly, M. M., and Houlden, H. (2023). “RFC1 CANVAS / Spectrum Disorder,” in GeneReviews®, eds. M. P. Adam, G. M. Mirzaa, R. A. Pagon, S. E. Wallace, L. J. Bean, K. W. Gripp, et al. (Seattle (WA): University of Washington, Seattle). Available at: http://www.ncbi.nlm.nih.gov/books/NBK564656/. (accessed on 10 August 2023).
- Cortese, A., Simone, R., Sullivan, R., Vandrovcova, J., Tariq, H., Yau, W. Y., et al. (2019). Biallelic expansion of an intronic repeat in RFC1 is a common cause of late-onset ataxia. Nat Genet 51, 649–658. [CrossRef]
- Cortese, A., Tozza, S., Yau, W. Y., Rossi, S., Beecroft, S. J., Jaunmuktane, Z., et al. (2020). Cerebellar ataxia, neuropathy, vestibular areflexia syndrome due to RFC1 repeat expansion. Brain 143, 480–490. [CrossRef]
- Currò, R., Dominik, N., Facchini, S., Vegezzi, E., Sullivan, R., Galassi Deforie, V., et al. (2024). Role of the repeat expansion size in predicting age of onset and severity in RFC1 disease. Brain, awad436. [CrossRef]
- Dominik, N., Magri, S., Currò, R., Abati, E., Facchini, S., Corbetta, M., et al. (2023). Normal and pathogenic variation of RFC1 repeat expansions: implications for clinical diagnosis. Brain 146, 5060–5069. 506. [CrossRef]
- Fonte, J., Machado, C., Oliveira, J., and Magalhães, M. (2023). Lower Facial Dystonia: An Unexpected Presentation Associated with Pathogenic RFC1 Repeat Expansions. Mov Disord Clin Pract 10, 1150–1151. [CrossRef]
- Ghorbani, F., De Boer-Bergsma, J., Verschuuren-Bemelmans, C. C., Pennings, M., De Boer, E. N., Kremer, B., et al. (2022). Prevalence of intronic repeat expansions in RFC1 in Dutch patients with CANVAS and adult-onset ataxia. J Neurol 269, 6086–6093. [CrossRef]
- Hadjivassiliou, M., Currò, R., Beauchamp, N., Dominik, N., Grunewald, R. A., Shanmugarajah, P., et al. (2023). Can CANVAS due to RFC1 biallelic expansions present with pure ataxia? J Neurol Neurosurg Psychiatry, jnnp-2023-331381. [CrossRef]
- Hirano, M., Kuwahara, M., Yamagishi, Y., Samukawa, M., Fujii, K., Yamashita, S., et al. (2023). CANVAS-related RFC1 mutations in patients with immune-mediated neuropathy. Sci Rep 13, 17801. [CrossRef]
- Kytövuori, L., Sipilä, J., Doi, H., Hurme-Niiranen, A., Siitonen, A., Koshimizu, E., et al. (2022). Biallelic expansion in RFC1 as a rare cause of Parkinson’s disease. NPJ Parkinsons Dis 8, 6. [CrossRef]
- Migliaccio, A. A., Halmagyi, G. M., McGarvie, L. A., and Cremer, P. D. (2004). Cerebellar ataxia with bilateral vestibulopathy: description of a syndrome and its characteristic clinical sign. Brain 127, 280–293. [CrossRef]
- Nakamura, H., Doi, H., Mitsuhashi, S., Miyatake, S., Katoh, K., Frith, M. C., et al. (2020). Long-read sequencing identifies the pathogenic nucleotide repeat expansion in RFC1 in a Japanese case of CANVAS. J Hum Genet 65, 475–480. [CrossRef]
- Rafehi, H., Szmulewicz, D. J., Bennett, M. F., Sobreira, N. L. M., Pope, K., Smith, K. R., et al. (2019). Bioinformatics-Based Identification of Expanded Repeats: A Non-reference Intronic Pentamer Expansion in RFC1 Causes CANVAS. The American Journal of Human Genetics 105, 151–165. [CrossRef]
- Ronco, R., Perini, C., Currò, R., Dominik, N., Facchini, S., Gennari, A., et al. (2023). Truncating Variants in RFC1 in Cerebellar Ataxia, Neuropathy, and Vestibular Areflexia Syndrome. Neurology 100, e543–e554. [CrossRef]
- Scriba, C. K. , Beecroft, S. J., Clayton, J. S., Cortese, A., Sullivan, R., Yau, W. Y., et al. (2020). A novel RFC1 repeat motif (ACAGG) in two Asia-Pacific CANVAS families. Brain, 2910. [Google Scholar] [CrossRef]
- Szmulewicz, D. J., Waterston, J. A., MacDougall, H. G., Mossman, S., Chancellor, A. M., McLean, C. A., et al. (2011). Cerebellar ataxia, neuropathy, vestibular areflexia syndrome (CANVAS): a review of the clinical features and video-oculographic diagnosis. Ann N Y Acad Sci 1233, 139–147. [CrossRef]
- The ACMG Laboratory Quality Assurance Committee, Richards, S., Aziz, N., Bale, S., Bick, D., Das, S., et al. (2015). Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 17, 405–423. [CrossRef]
- Tsuchiya, M., Nan, H., Koh, K., Ichinose, Y., Gao, L., Shimozono, K., et al. (2020). RFC1 repeat expansion in Japanese patients with late-onset cerebellar ataxia. J Hum Genet 65, 1143–1147. [CrossRef]
- Wu, T. Y., Taylor, J. M., Kilfoyle, D. H., Smith, A. D., McGuinness, B. J., Simpson, M. P., et al. (2014). Autonomic dysfunction is a major feature of cerebellar ataxia, neuropathy, vestibular areflexia “CANVAS” syndrome. Brain 137, 2649–2656. [CrossRef]
Figure 1.
The proposed CANVAS testing approach and interpretation. RT-PCR, repeat-primed PCR; P/LP SNV, pathogenic/likely pathogenic single nucleotide variants; ES, exome sequencing.
Figure 1.
The proposed CANVAS testing approach and interpretation. RT-PCR, repeat-primed PCR; P/LP SNV, pathogenic/likely pathogenic single nucleotide variants; ES, exome sequencing.
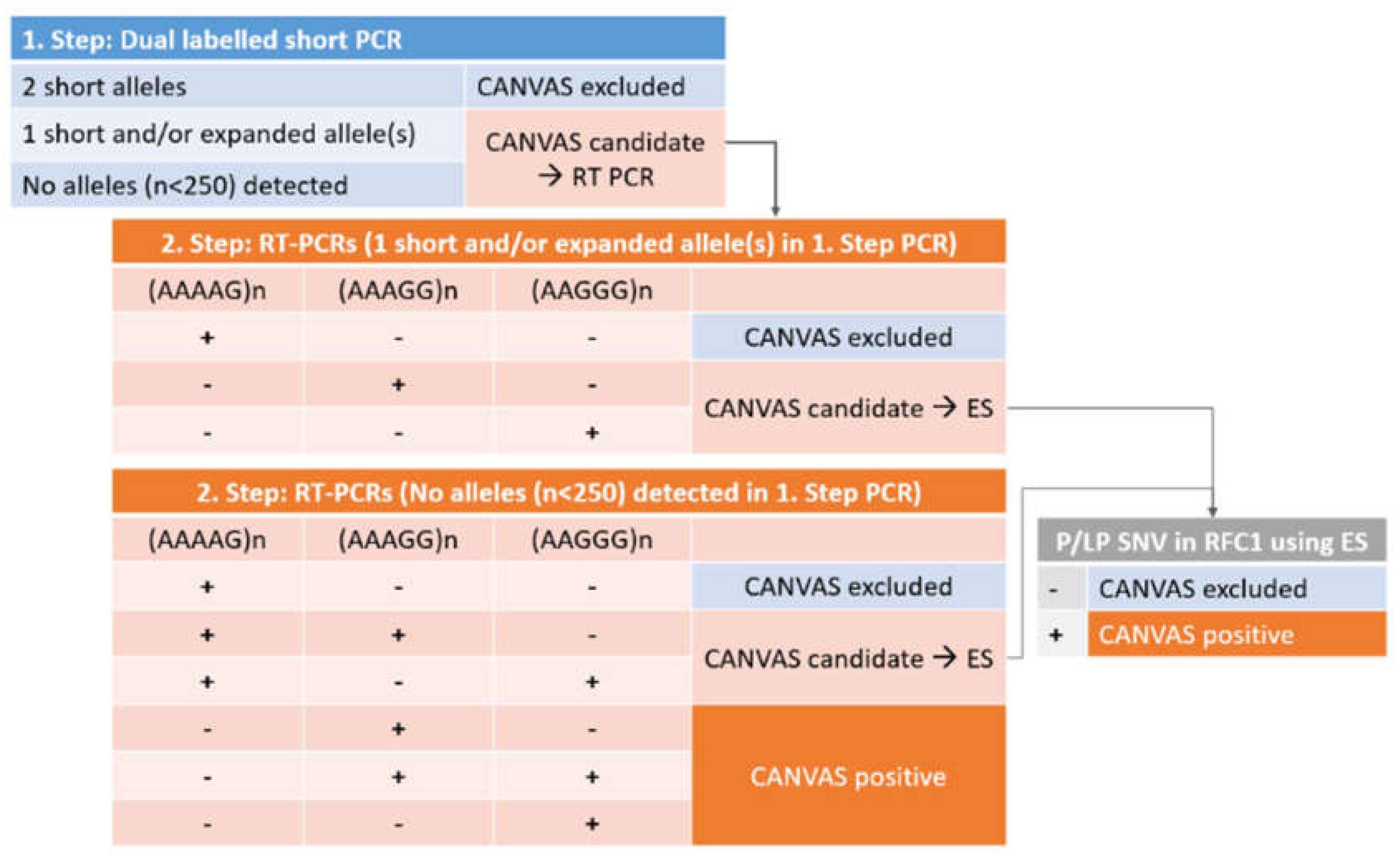
Figure 2.
Two-step PCR and fragment analysis principle.
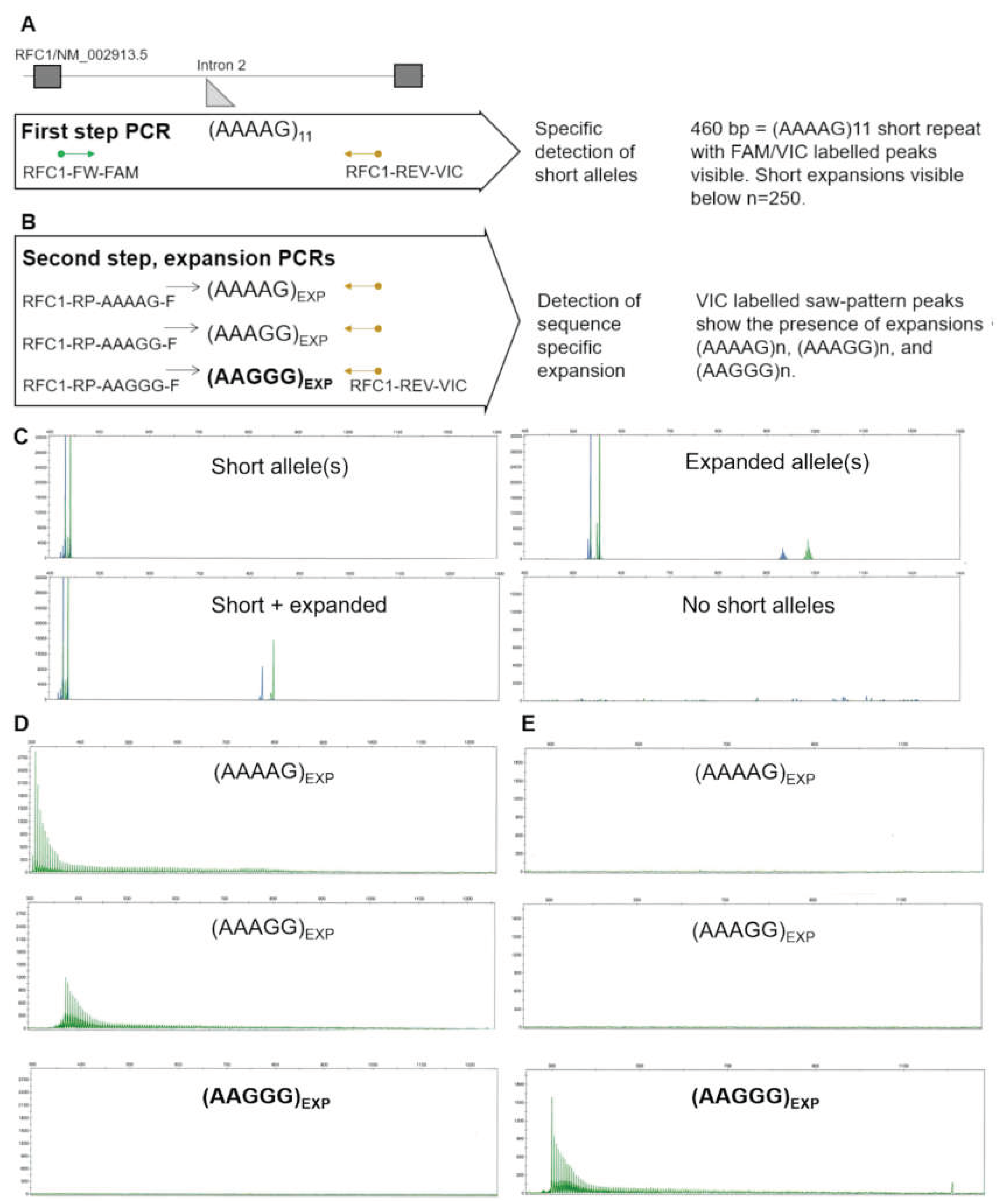

Table 1.
Results of two-step PCR testing in patients with known non-CANVAS genetic cause of ataxia.
| Patients, N | First-step PCR | Patients with confirmatory PCR | Final result (%) | ||
|---|---|---|---|---|---|
| AAAAG | AAAGG | AAGGG | |||
| 6 | 2 short alleles | 0 | 0 | 0 | Negative |
| 8 | 1 short allele | 6 | 0 | 0 | |
| 0 | 0 | 0 | |||
| 1 | 1 short, 1 expanded allele |
1 | 0 | 0 | |
| 2 | 1 expanded allele | 2 | 0 | 0 | |
| 2 | 2 expanded alleles | 2 | 0 | 0 | |
| Total | 19 (100.0%) | ||||
| 0 | No short alleles | Positive | |||
| Total | 0 (0.0%) | ||||
| Total | 11 | 0 | 0 | ||
Table 2.
Results of two-step PCR testing in consecutively referred patients with clinically suspected CANVAS.
Table 2.
Results of two-step PCR testing in consecutively referred patients with clinically suspected CANVAS.
| Patients, N | First-step PCR | Patients with confirmatory PCR | Final result (%) | ||
|---|---|---|---|---|---|
| AAAAG | AAAGG | AAGGG | |||
| 2 | 2 short alleles | 0 | 0 | 0 | Negative |
| 2 | 1 short allele | 1 | 0 | 0 | |
| 0 | 0 | 0 | |||
| 0 | 1 short, 1 expanded allele |
0 | 0 | 0 | |
| 1 | 1 expanded allele | 1 | 0 | 0 | |
| 3 | 2 expanded alleles | 3 | 0 | 0 | |
| 7 | No short alleles | 0 | 0 | 0 | |
| 1 | 1 | 0 | |||
| 0 | 0 | 0 | |||
| Total | 10 (66.6 %) | ||||
| 0 | 0 | 5 | Positive | ||
| Total | 5 (33.3 %) | ||||
| Total | 6 | 1 | 5 | ||
Table 3.
Clinical characteristics and genetic testing results of Slovenian patients with suspected CANVAS.
Table 3.
Clinical characteristics and genetic testing results of Slovenian patients with suspected CANVAS.
| Patient ID | Age of onset | Sex | Cerebellar ataxia | Bilateral vestibular areflexia | Sensory neuropathy/neuronopathy | Other symptoms | CANVAS testing result |
|---|---|---|---|---|---|---|---|
| P01 | 53 | M | + | + | + | - | Confirmed, + Southern blot |
| P02 | 39 | F | + | + | + | autonomic dysfunction | Confirmed, + Southern blot |
| P03 | 53 | M | + | n.d. | + | autonomic dysfunction, hearing loss | Confirmed, + Southern blot |
| P04 | n.d. | M | + | n.d. | + | autonomic dysfunction, hearing loss | Confirmed |
| P05 | 61 | M | + | + | + | - | Confirmed |
| P06 | 55 | M | + | malfunction of the right vestibular apparatus | + | chronic cough | Confirmed |
| P07 | 50 | F | + | + | + | autonomic dysfunction | Confirmed |
| P08 | 58 | F | + | + | + | - | Confirmed |
| P09 | 68 | M | + | - | + | hearing loss | Negative * |
| P10 | 27 | F | + | - | - | hearing loss | Negative **,# |
| P11 | 20 | F | + | - | - | hearing loss, cervical segmental dystonia | Negative |
| P12 | n.d. | M | + | n.d. | - | - | Negative *** |
| P13 | 40 | M | + | n.d. | - | - | Negative *** |
| P14 | n.d | F | + | n.d. | - | - | Negative *** |
| P15 | 17 | M | + | n.d. | + | spastic paraparesis | Negative *** |
| P16 | 55 | M | + | n.d. | - | parkinsonism, autonomic dysfunction | Negative |
| P17 | 70 | F | + | n.d. | - | - | Negative |
| P18 | 68 | F | + | n.d. | + | - | Negative |
* patient has no short allele, and none of the three most common expansion sequence motifs, ** Patient has both (AAAAG)n and (AAAGG)n expansions, # Exome sequencing performed, no pathogenic single nucleotide variants identified in RFC1, *** patient has both alleles expanded for (AAAAG)n non-pathogenic repeats, n.d. no data.
Table 4.
Primers used for dual-labelled short PCR (screening) and RT-PCR (confirmatory) PCRs.
| Primer | Sequence (5’ - 3’) | Short PCR | RT-PCRs |
|---|---|---|---|
| RFC1-FW-FAM | FAM-CTGAGGCAGGAGATTCACTTG | Fw PCR1 | |
| RFC1-REV-VIC | VIC-GGTGGCTGTCTCATCTGTTG | Rev PCR1 | Rev PCR 2, 3, 4 |
| RFC1-RP-AAAAG-F | GAAAAGAAAAGAAAAGAAAAGAAAAG | Fw PCR 2 | |
| RFC1-RP-AAAGG-F | AAAGGAAAGGAAAGGAAAGG | Fw PCR 3 | |
| RFC1-RP-AAGGG-F | GAAGGGAAGGGAAGGGAA | Fw PCR 4 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



