Submitted:
10 November 2020
Posted:
10 November 2020
You are already at the latest version
Abstract
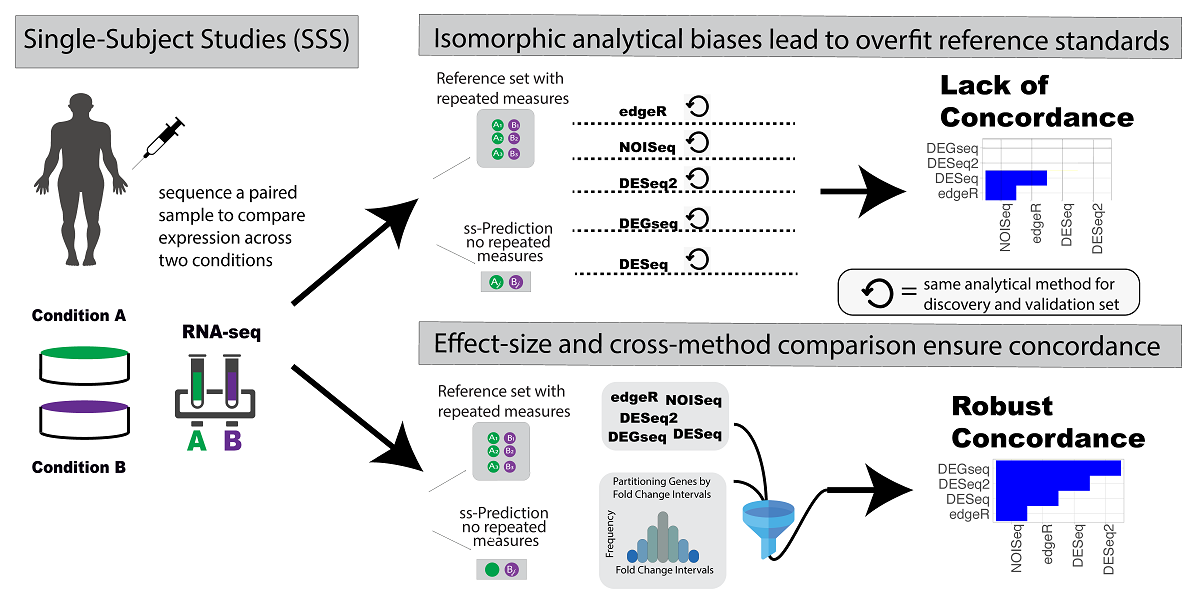
Background: Developing patient-centric baseline standards that enable the detection of clinically significant outlier gene products on a genome-scale remains an unaddressed challenge required for advancing personalized medicine beyond the small pools of subjects implied by “precision medicine”. This manuscript proposes a novel approach for reference standard development to evaluate the accuracy of single-subject analyses of metabolomes, proteomes, or transcriptomes. Since distributional assumptions of statistical testing may inadequately model genome dynamics of gene products, the so-called significant results of previous studies may artefactually conflate with real signals. Model confirmation biases escalate when studies use the same analytical methods in the discovery sets and reference standards, as corroboration of results leads to an evaluation of reproducibility confounded with replicated biases rather than a measure of accuracy. We hypothesized that developing method-agnostic reference standards using effect-size and expression-level filtering of results, obtained from multiple discovery methods that are distinct from the one evaluated, would maximize the evaluation of clinical-transcriptomic signals and minimize statistical artefactual biases. We developed and released an R package “referenceNof1” to facilitate the construction of robust reference standards. Results: Since RNA-Seq data analysis methods often rely on binomial and negative binomial assumptions to non-parametric analyses, the differences create statistical noise and make the reference standards method dependent. In our experimental design, the accuracy of 30 distinct combinations of fold changes (FC) and expression levels (EL) were determined for five types of RNA analyses in two different datasets. This design was applied to two distinct datasets: breast cancer cell lines and a yeast study with isogenic biological replicates in two experimental conditions. In addition, the reference standard (RS) comprised all RNA analytical methods with the exception of the method testing accuracy. To mitigate for biased optimization of the RS parameters towards a specific analytical method, similarity between observed results of distinct analytical methods were calculated across all methods (Jaccard Concordance Index). The greatest differences were observed across diametric extremes. For example, filtering out differentially expressed genes (DEGs) using a fold change < 1.2 leads to a 50% increase in concordance between techniques when compared to results with FC > 1.2. Combining this FC cutoff with genes with mean expressions > 30 counts leads to a 65% increase in concordance in comparison to genes with expression levels < 30 counts and with FC < 1.2. Conclusions: We have demonstrated that comparing accuracies of different single-subject analysis methods for clinical optimization requires a new evaluation framework. Reliable and robust reference standards, independent of the evaluated method, can be obtained under a limited number of parameter combinations: fold change (FC) ranges thresholds, expression level cutoffs, and exclusion of the tested method from the RS development process. When applying anticonservative reference standard frameworks (e.g., using the same method for RS development and for prediction), a majority of the concordant signal between prediction and Gold Standard (GS) cannot be confirmed by other methods, which we conclude as biased results. Statistical tests to determine DEGs from a single-subject study generate many biased results that require subsequent filtering for increasing their reliability. Conventional single-subject studies pertain to one or a few measures in one patient over time [1]and need a substantial conceptual framework extension in order to address the tens of thousands of measures in genome-wide analyses of gene products. The proposed referenceNof1 framework addresses some of the inherent challenges in improving transcriptome scale single-subject analyses by providing a robust approach to constructing reference standards. Github: https://github.com/SamirRachidZaim/referenceNof1
Keywords:
single-subject studies
; personalized medicine
; precision medicine
; reference standards
; gold standards
; biomarkers
; open-source
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



