Submitted:
20 May 2025
Posted:
20 May 2025
You are already at the latest version
Abstract
Lysosome is crucial for maintaining cellular homeostasis, but disintegrity of its limiting membrane affects the cell death fate. From 1972 to 1999, via the cytochemistry of cultured cells which were exposed to stresses, Brunk et al. defined lysosomal membrane permeabilization (LMP) as leakage through the ultrastructurally-intact limiting membrane. In 1996, via the electron microscopic analysis of the monkey hippocampal CA1 neurons after transient ischemia, Yamashima et al. identified lysosomal membrane rupture (LMR) as an apparent disruption of the limiting membrane. To elucidate the mechanism of lysosomal cell death, it is indispensable to precisely differentiate LMP and its extensive form LMR. LMP indicates formation of ultrastructurally-undetectable, tiny pores at the lysosomal limiting membrane that allow selective leakage of lysosomal contents. LMP contributes to amplification of the cell death signal, and participates in apoptosis. If only a small number of lysosomes is affected, the cell can eliminate injured lysosomes via activating lysophagy, and ensure cell survival. However, when majority of lysosomes are affected, the cell develops apoptosis. In contrast, LMR indicates presence of larger holes that cause acute and massive leakage of hydrolytic cathepsin enzymes. LMR leads to the rapid and explosive vanishment of lysosomes, which results in necrosis. Each form of cell death is carried out, depending upon the size and number of perforations, the amount of leakage, and the cellular context. The modality of the lysosomal membrane disintegrity, i.e., LMP or LMR, determines cell death fate. It is likely that apoptosis occurs by the proteolytic activation of caspases via LMP, whereas necrosis occurs by the calpain-cathepsin cascade via LMR. This paper is to review ultrastructural evidences of LMR which were identified in diverse pathologic conditions of C. elegans, mice, monkeys, and humans. For elucidating mechanisms of each cell death at stresses or diseases, LMP and LMR should be precisely differentiated by electron microscopy.
Keywords:
lysosome
; cell death
; hydroxynonenal
; apoptosis
; necrosis
1. Introduction
Lysosomes are membrane-bound vesicular structures which contain more than 60 acid hydrolytic enzymes including proteases, phosphatases, nucleases, glycosidases, peptidases, sulphatases, and lipases [de Duve, 1983]. Their main function is to degrade macromolecules being internalized by endocytosis/phagocytosis and intercellular aged/damaged components. Hydrolytic enzymes break down complex macromolecules into their building blocks or amino acids within the lysosomal lumen for recycling. Since de Duve discovered lysosomes in 1955 [1], researchers made significant contributions to link lysosomes with cell death of eukaryotic cells. Christian de Duve was awarded Nobel prize in 1974 for the discovery of lysosomes. For ensuring the place of degradation within the acidic lumen at ~pH4.5, the integrity of lysosomal limiting membranes is critical in order not to damage the cell via the leakage of hydrolytic enzymes into the cytoplasm. For this purpose, the lysosomal limiting membrane is approximately 8 nm thick, and has a highly-glycosylated transmembrane protein such as lysosome-associated membrane proteins 2 (LAMP-2). Both protect the membrane from inside not to be degraded by its own hydrolytic enzymes [2–4].
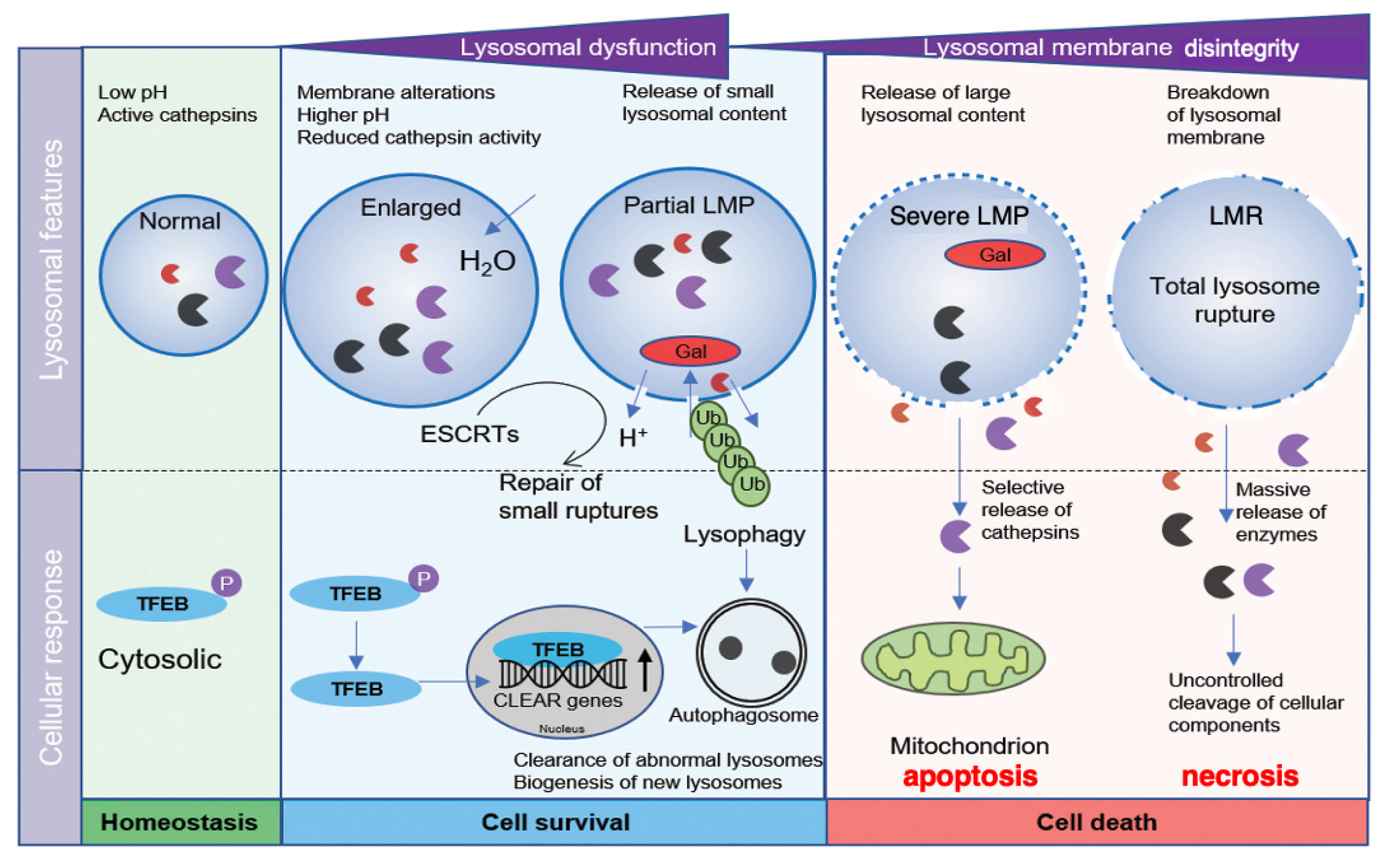
In normal conditions, lysosomal transcription factor EB (TFEB) is phosphorylated in the cytosol [Figure 1, light-green]. At the lysosomal stress, TFEB is dephosphorylated and translocates to the nucleus, where it upregulates the transcription of coordinated lysosomal expression and regulation (CLEAR) genes. CLEAR genes encode hydrolases, lysosomal membrane proteins, and the v-ATPase proton pump complex, participating in the autophagosome formation [5]. If the lysosomal membrane disintegrity is partial or if only a small subset of lysosomes is affected at the cellular stresses, the regulation by lysophagy and TFEB is initiated to ensure cell survival. Lysophagy senses pores being permeable to galectins at the lysosomal limiting membrane. The endosomal sorting complex required for transport proteins (ESCRT) can detect and repair small pores in the membrane [6] [Figure 1, light-blue]. In cases of more severe lysosomal disintegrity, however, such stress response mechanisms are unable to repair larger perforations, and result in cell death [7] [Figure 1, light-orange]. Over the last three decades, the lysosome has emerged as an important executor of the cell death machinery.
Figure 1.
Relation between the lysosomal membrane integrity/disintegrity and cell survival/death. TFEB is phosphorylated in the cytosol in the normal state (light-green). At the partial LMP (light-blue), stress-sensing and response mechanisms including lysophagy and TFEB regulation, are initiated to ensure cell survival. In cases of more severe LMP (light-orange), the stress response mechanisms are unable to repair damaged lysosomes. Selective release of cathepsins by the severe LMP (light-orange) results in mitochondrial damage and apoptosis. In the most severe situations of LMR (light-orange), rupture of the lysosomal limiting membrane causes the massive release of lysosomal cathepsin enzymes, uncontrolled breakdown of cell constitutive proteins, and necrosis. CLEAR, coordinated lysosomal expression and regulation; ESCRT, endosomal sorting complex required for transport; Gal, galectin; LMP, lysosomal membrane-permeabilization; LMR, lysosomal membrane-rupture; TFEB, transcription factor EB; Ub, ubiquitin. (Cited and adapted from [7]).
Figure 1.
Relation between the lysosomal membrane integrity/disintegrity and cell survival/death. TFEB is phosphorylated in the cytosol in the normal state (light-green). At the partial LMP (light-blue), stress-sensing and response mechanisms including lysophagy and TFEB regulation, are initiated to ensure cell survival. In cases of more severe LMP (light-orange), the stress response mechanisms are unable to repair damaged lysosomes. Selective release of cathepsins by the severe LMP (light-orange) results in mitochondrial damage and apoptosis. In the most severe situations of LMR (light-orange), rupture of the lysosomal limiting membrane causes the massive release of lysosomal cathepsin enzymes, uncontrolled breakdown of cell constitutive proteins, and necrosis. CLEAR, coordinated lysosomal expression and regulation; ESCRT, endosomal sorting complex required for transport; Gal, galectin; LMP, lysosomal membrane-permeabilization; LMR, lysosomal membrane-rupture; TFEB, transcription factor EB; Ub, ubiquitin. (Cited and adapted from [7]).
Regardless of insults, cell types, organs, diseases, or species, leakage of the lysosomal content occurs via the disruption of the lysosomal limiting membrane. There are essentially two types of the lysosomal membrane disintegrity: the ultrastructurally-intact lysosomal limiting membrane showing permeabilization via formation of tiny pores (lysosomal membrane-permeabilization, LMP), or the massive leakage of the lysosomal content through the apparent rupture (larger holes) of the lysosomal limiting membrane (lysosomal membrane-rupture, LMR) [Figure 1, light-orange] [4]. In response to acute intense insults, lysosomes fade away via the explosion-like rupture. Therefore, dissolving lysosomes are hardly detected during the phase of cell degeneration, and lysosomes in dying cells had been less frequently focused by the previous investigators [8].
By focusing on changes of the lysosomal membrane integrity with the time-lapse imaging and electron microscopic analyses, herein, implications of LMP/LMR for the cell apoptosis/necrosis were discussed.
2. Permeabilization or Rupture of Lysosomal Limiting Membranes
More than half century ago, de Duve and Wattiaux proposed such a concept that cell degeneration and necrosis occur in pathological states via the leakage of hydrolytic enzymes from damaged lysosomes [9]. Concerning the lysosomal leakage, it is likely that LMP induces the gradual and selective release of hydrolytic cathepsin enzymes, whereas LMR induces the rapid and massive release of cathepsins (Figure 1, light-orange). The main questions to be addressed are 1) how does disintegrity of the lysosomal limiting membranes occur?, 2) what specifically occurs after the formation of pores or holes at the lysosomal limiting membranes?, 3) how do pores or holes participate in the execution of the peculiar cell death, and 4) how are they identified in the process of cell degeneration?.
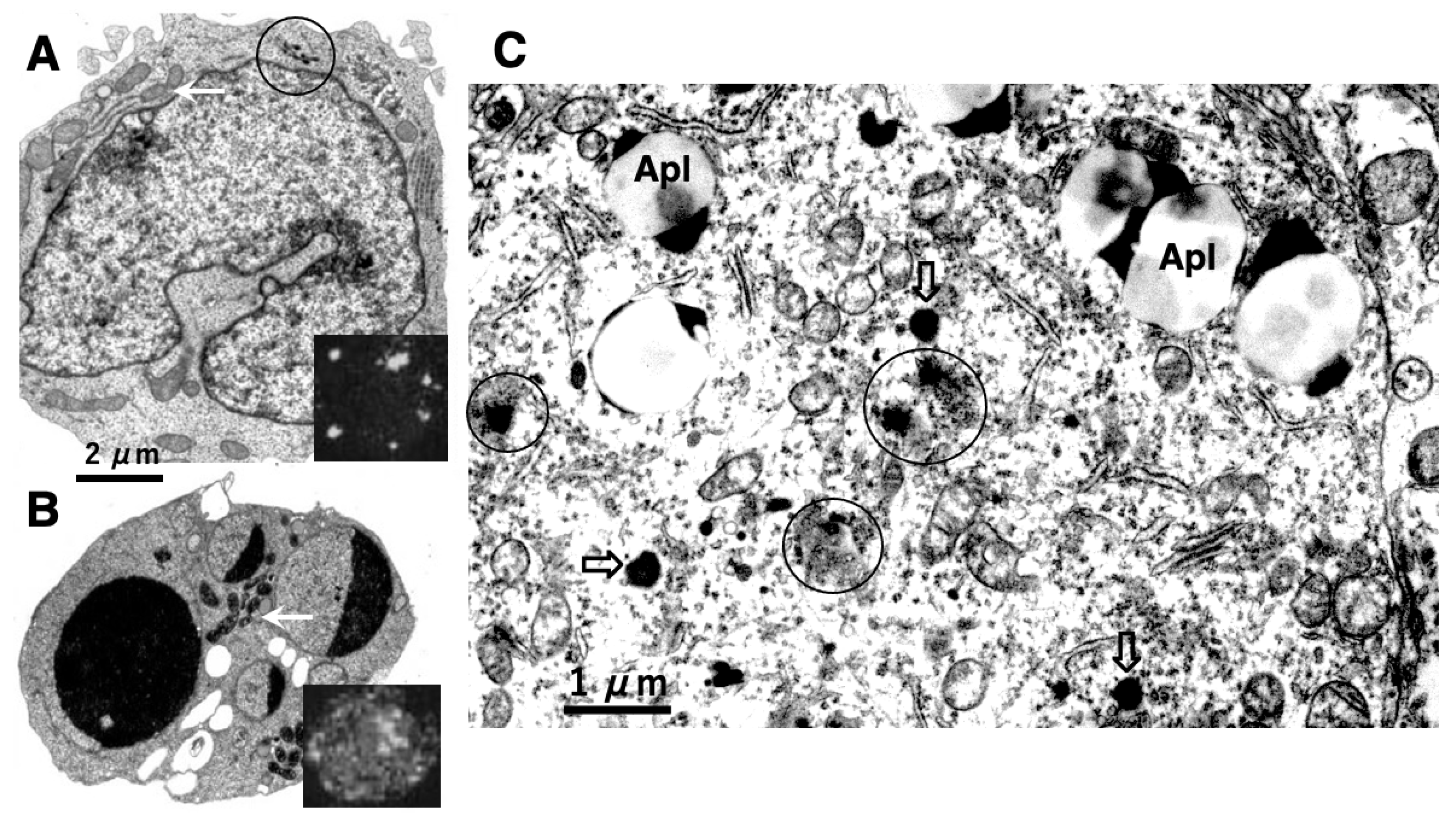
To address these issues, it is indispensable to elucidate whether cell degeneration/death occurs via ultrastructurally-intact lysosomes. In 1972, Brunk and Ericsson found by cytochemistry an extensive diffusion of lysosomal acid phosphatases through the ultrastructurally-intact lysosomal limiting membrane, using the cultured human glioma cells after the fixation at an improper osmotic pressure [10]. Presumably, pores allowing translocation of hydrolytic enzymes were formed artificially at the lysosomal limiting membranes by the improper fixation. In 1999, using a human T-leukemia cell line, Jurkat EG.1 cells, which were exposed to oxidative stresses, Brunk and Svensson first confirmed implication of LMP, i.e., leakage of acridine orange through the lysosomal limiting membrane [11] (Figure 2B, insert), which was not seen in the control (Figure 2A, insert). As escaping detection from the electron microscopic observation, Brunk and his colleagues speculated implication of tiny pores at the lysosomal limiting membranes, i.e., LMP, for the development of apoptosis (Figure 2B) [11–14].
Figure 2.
Electronmicrophotographs of Jurkat T-leukemia cells (A,B) and human cortical neuron of Alzheimer’s disease (C). The control Jurkat T-leukemia cell (A) is characterized by a folded nucleus with diffuse chromatin distribution, normal mitochondria (arrow), and a few lysosomes (circle). Following exposure to the anti-human Fas/APO-1/CD95 antibody, however, evidence of apoptosis such as nuclear chromatin condensation, fragmentation, and capping was seen (B). An increase of the mitochondrial electron density was confirmed after the insult (B, arrow), as compared to the control (A, arrow). Lysosomal membrane disintegrity was not detected by electron microscopy. By light microscopy, however, cells treated with acridine orange following exposure to the antibody show leakage of dye throughout the cytoplasm (B, insert), showing a marked contrast to the control (A, insert) These data indicated implication of LMP, but not of LMR, in the occurrence of apoptosis. (Cited from [11]). In the human brain also, leakage of the lysosomal content was observed in the cortical neuron of Alzheimer’s disease (C, circles). As compared to the normal lysosomes (open arrows), many lysosomes are not bound by distinct membranes, showing apparent leakage of the lysosomal contents by LMR (circles). This showed a marked contrast to T-leukemia cells which exhibited LMP, but not LMR. Apl; autophagolysosomes. (Cited from [15]).
Figure 2.
Electronmicrophotographs of Jurkat T-leukemia cells (A,B) and human cortical neuron of Alzheimer’s disease (C). The control Jurkat T-leukemia cell (A) is characterized by a folded nucleus with diffuse chromatin distribution, normal mitochondria (arrow), and a few lysosomes (circle). Following exposure to the anti-human Fas/APO-1/CD95 antibody, however, evidence of apoptosis such as nuclear chromatin condensation, fragmentation, and capping was seen (B). An increase of the mitochondrial electron density was confirmed after the insult (B, arrow), as compared to the control (A, arrow). Lysosomal membrane disintegrity was not detected by electron microscopy. By light microscopy, however, cells treated with acridine orange following exposure to the antibody show leakage of dye throughout the cytoplasm (B, insert), showing a marked contrast to the control (A, insert) These data indicated implication of LMP, but not of LMR, in the occurrence of apoptosis. (Cited from [11]). In the human brain also, leakage of the lysosomal content was observed in the cortical neuron of Alzheimer’s disease (C, circles). As compared to the normal lysosomes (open arrows), many lysosomes are not bound by distinct membranes, showing apparent leakage of the lysosomal contents by LMR (circles). This showed a marked contrast to T-leukemia cells which exhibited LMP, but not LMR. Apl; autophagolysosomes. (Cited from [15]).
As LMP did not change the ultrastructure of lysosomes [10], the interests in lysosomes, lysosomal membrane disintegrity and the resultant cell death gradually faded, and lysosomal involvement in cell death was overlooked thereafter. For a long time, the lysosome had been imprecisely considered a sturdy organelle that shows increased permeability, but does not show apparent rupture until the cell is already devitalized [16]. It was believed that massive leakage of hydrolytic enzymes from injured lysosomes, if occurs, would be a late event in the terminal phase of cell degeneration or as the postmortal alteration in autolytic cells.
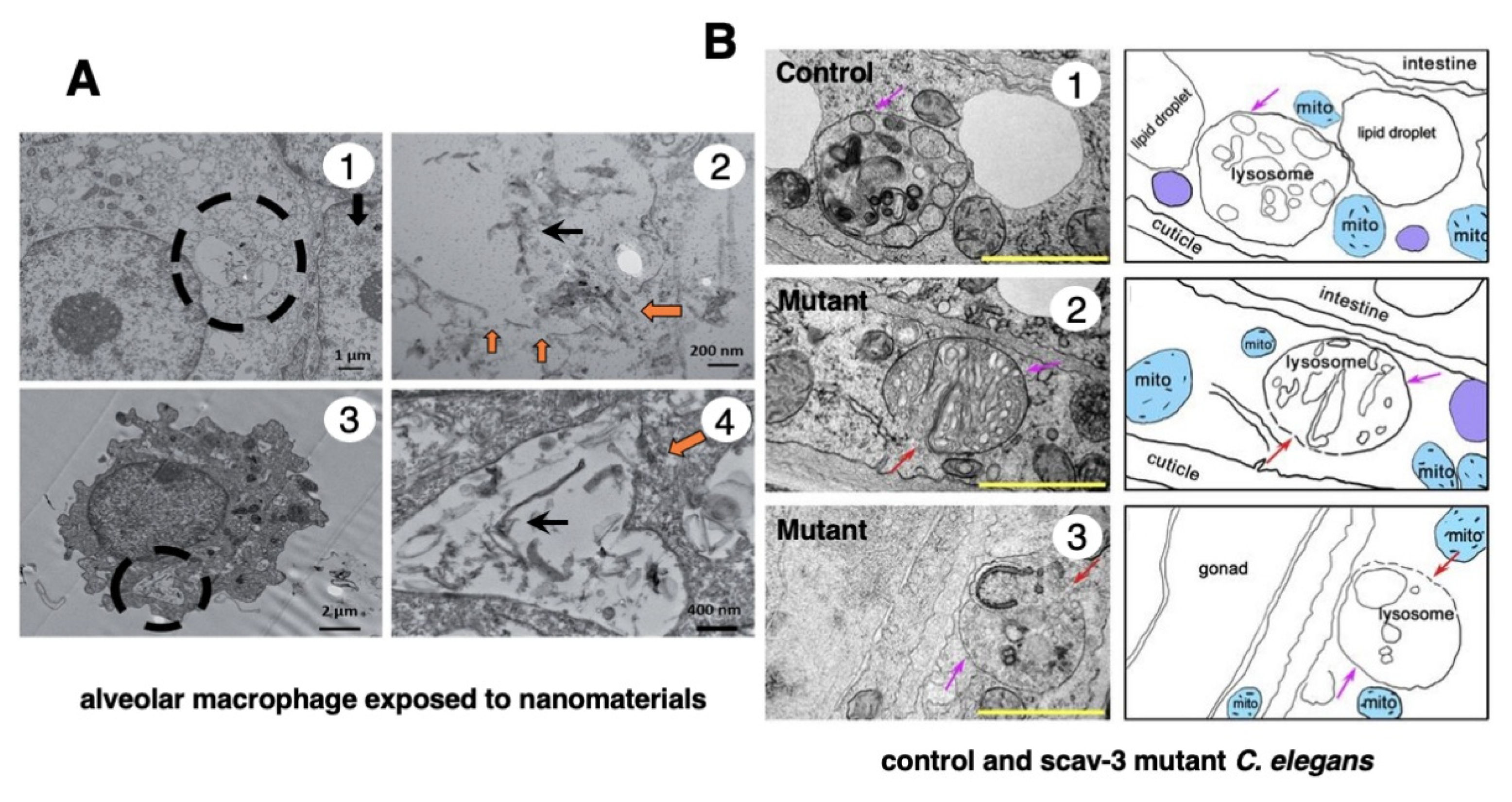
In 1996, however, in the degenerating, but still alive, hippocampal cornu Ammonis 1 (CA1) neurons of Japanese macaque monkeys on days 3-5 after transient, global brain ischemia, Yamashima et al. found by electron microscopy evidence of LMR forming larger holes which allow leakage of a massive amount of the lysosomal contents (Figure 3B, arrows), prior to the execution of necrosis [8]. It showed a remarkable contrast to the normal lysosomes (Figure 3A, circles) in the non-ischemic CA1 neurons. Later, the similar LMR was confirmed in the phagolysosomes of both the cultured macrophages and alveolar macrophages of mice which were exposed to nanomaterials (Figure 4A, orange arrows) [17]. SCAV-3 is the C. elegans homologue of human lysosomal integral membrane protein type 2 (LIMP-2, also known as SCARB2) which serves as one of the key regulators of lysosomal membrane integrity. Li et al. also found that the loss of the scav-3 gene in C. elegans causes LMR of the lysosomal limiting membranes (Figure 4B, red arrows) [18]. Intriguingly, Yamashima and his colleagues confirmed presence of the similar leakage of the lysosomal contents in the degenerating cortical neuron of patients with Alzheimer’s disease (Figure 2C, circles). By the careful observation, double contour of lysosomes indicating LMR was not infrequently detected in the vicinity of autophagolysosomes and degenerated mitochondria [15,19].
Figure 3.
Electronmicrophotographs of the monkey hippocampal CA1 neuron before (A) and after (B) the transient ischemia. The non-ischemic CA1 neuron (A) shows normal lysosomes (circles). In contrast, the CA1 neuron after the transient ischemia shows apparent disruptions, i.e., LMR (small arrows) of the lysosomal limiting membrane with apparent leakage (large arrows) of the lysosomal content (L). Mitochondria (m) show marked disruptions of inner membranes, while rough ER (stars) show swelling. The synaptic vesicles (B, open arrows) are decreased, compared to the control (A). N, nucleus; P, peroxisome. (cited from [8]).
Figure 3.
Electronmicrophotographs of the monkey hippocampal CA1 neuron before (A) and after (B) the transient ischemia. The non-ischemic CA1 neuron (A) shows normal lysosomes (circles). In contrast, the CA1 neuron after the transient ischemia shows apparent disruptions, i.e., LMR (small arrows) of the lysosomal limiting membrane with apparent leakage (large arrows) of the lysosomal content (L). Mitochondria (m) show marked disruptions of inner membranes, while rough ER (stars) show swelling. The synaptic vesicles (B, open arrows) are decreased, compared to the control (A). N, nucleus; P, peroxisome. (cited from [8]).
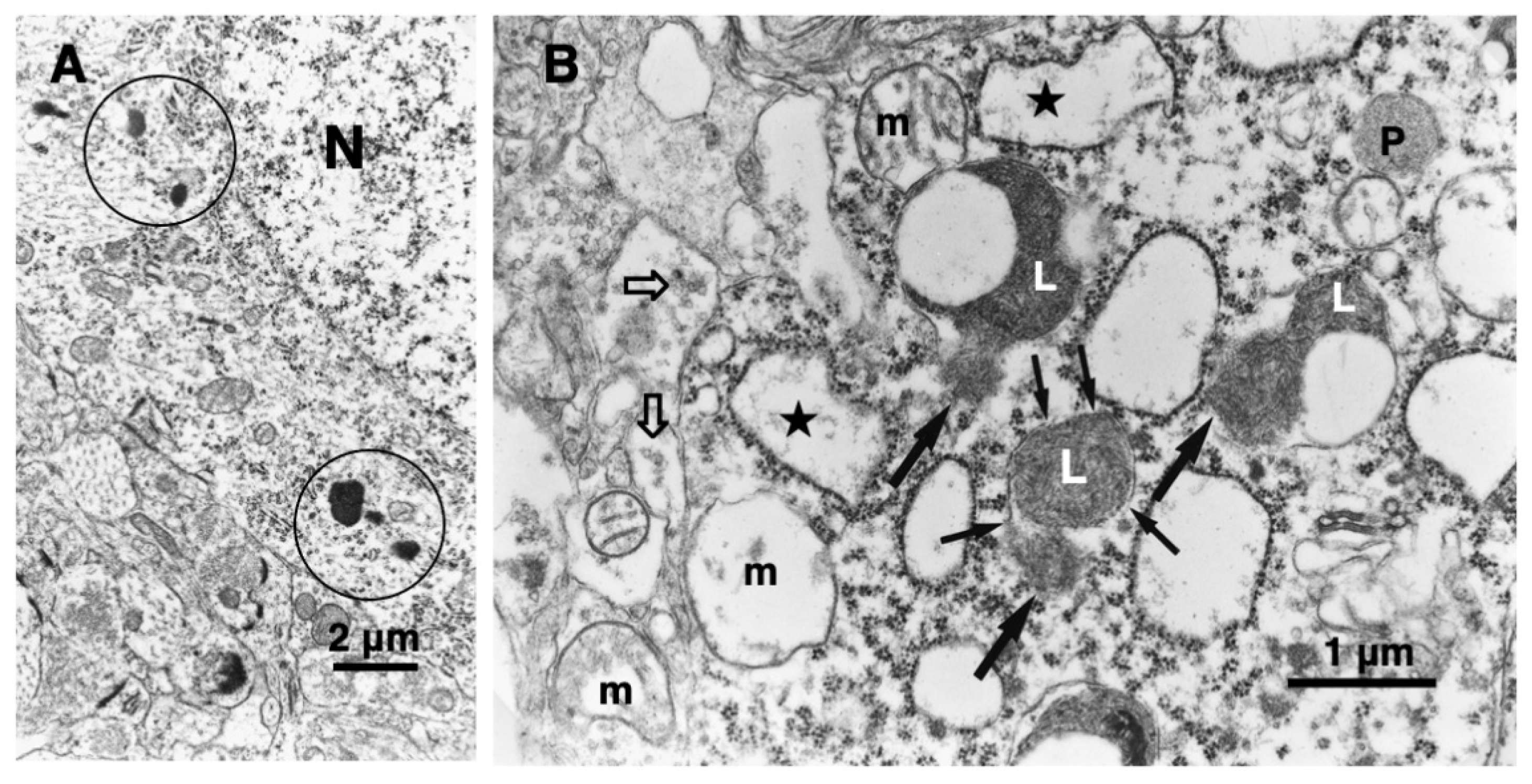
Figure 4.
Ultrastructural evidence of LMR which was confirmed in the cultured mice macrophage (A) and the scav-3 mutant C. elegans (B). The cultured alveolar macrophage exposed to nanomaterials (A-2,4, black arrows) show evidence of LMR (A-2,4, orange arrows) (Cited from [17]). SCAV-3 is the C. elegans homologue of human lysosomal integral membrane protein type 2 (LIMP-2, also known as SCARB2) which serves as one of the key regulators of lysosomal membrane integrity. The loss of the scav-3 gene in C. elegans causes rupture of the lysosomal limiting membranes (B-2,3, red arrows). Purple arrows indicate intact portion of the lysosomal limiting membrane. LMR in the cultured cell and C. elegans is very similar to LMR found in the monkey (Figure 3B). (Cited from [18]).
Figure 4.
Ultrastructural evidence of LMR which was confirmed in the cultured mice macrophage (A) and the scav-3 mutant C. elegans (B). The cultured alveolar macrophage exposed to nanomaterials (A-2,4, black arrows) show evidence of LMR (A-2,4, orange arrows) (Cited from [17]). SCAV-3 is the C. elegans homologue of human lysosomal integral membrane protein type 2 (LIMP-2, also known as SCARB2) which serves as one of the key regulators of lysosomal membrane integrity. The loss of the scav-3 gene in C. elegans causes rupture of the lysosomal limiting membranes (B-2,3, red arrows). Purple arrows indicate intact portion of the lysosomal limiting membrane. LMR in the cultured cell and C. elegans is very similar to LMR found in the monkey (Figure 3B). (Cited from [18]).
Nowadays, it is believed that a low level of cell stresses causes LMP and apoptosis by mitochondrial transmembrane potential loss or caspase activation [20–22]. In contrast, necrosis is triggered via LMR by more intense, catastrophic events such as heat shock, ischemia, irradiation, or irreparable oxidative stress to the cell [8,12,14,16,23–28]. In the consecutive works independently done by Brunk et al. and Yamashima et al., it was established that the extent of lysosomal disintegrity determines the cell death fate, i.e., a selective release of lysosomal contents results in apoptosis (Figure 2B) [10–14,29], whereas a massive lysosomal breakdown leads to necrosis [4,8,23,25,30,31] (Figure 7E).
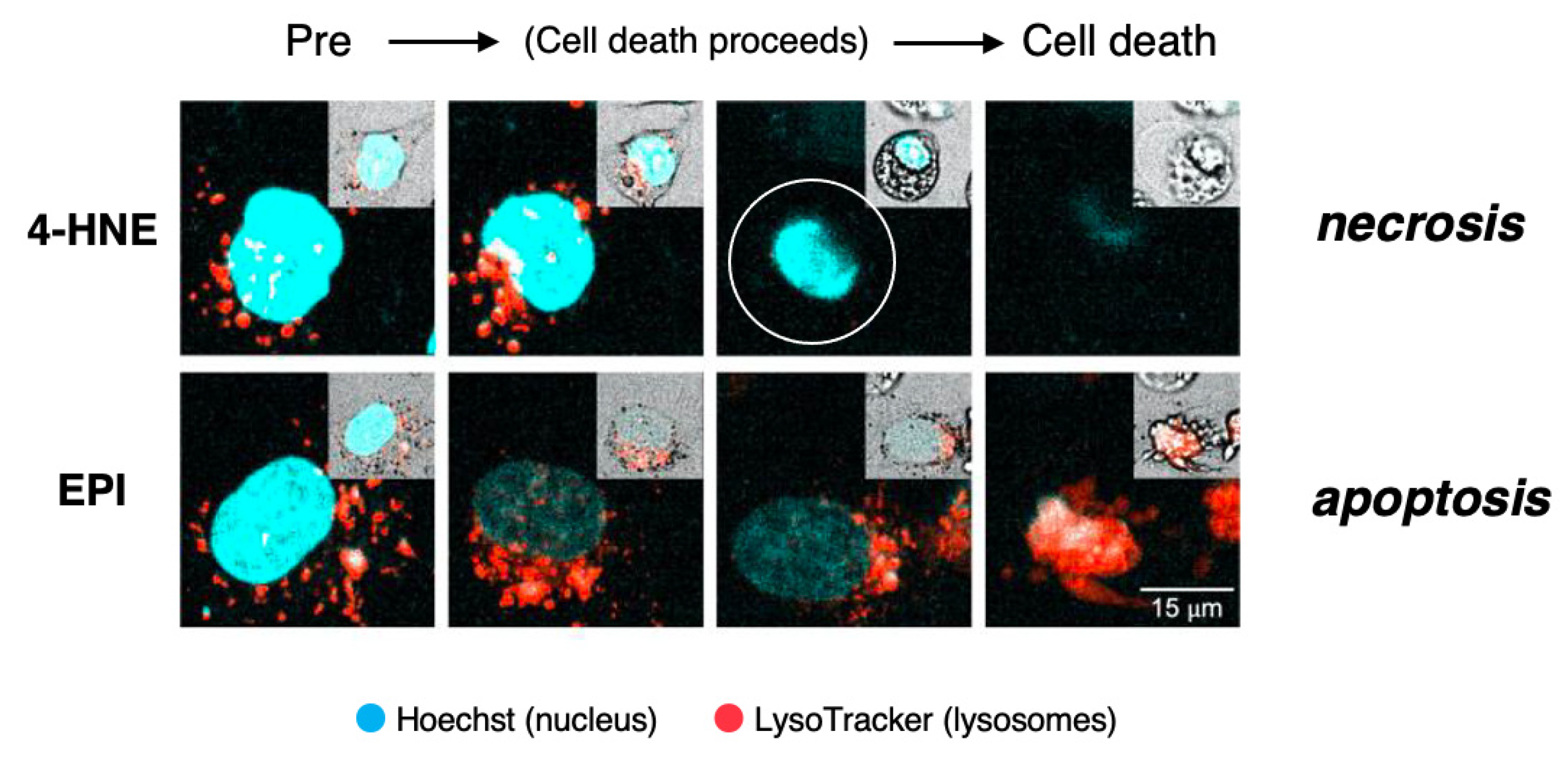
This concept was recently confirmed by us in the cultured hepatocellular carcinoma cell line HepG2 which was exposed to lipid-peroxidation product, 4-hydroxy-2-nonenal (4-HNE), as compared to treatment with antibiotic chemotherapeutic agent, epirubicin (EPI) [32] (Figure 5). By the time-lapse imaging using LysoTracker (as shown by orange color), which is a highly soluble small molecule that is retained in the acidic lysosomal compartment, induction of the lysosomal membrane disintegrity was studied.
The time-lapse imaging revealed that the addition of EPI to the HepG2 culture caused cell shrinkage and formation of blebbing with most of the lysosomes preserved until the execution of apoptosis. As the lysosomal limiting membranes remained ultrastructurally intact, it is likely that gradual and selective leakage had occurred presumably through tiny pores in the limiting membrane. Although acridine orange showed extralysosomal leakage, it grossly remained within lysosomes until apoptosis is completed by EPI. The combined immunoreactivity of acridine orange within lysosomes and perilysosomal area was enlarged, as compared to the control lysosomes (Figure 5, lower column). Although lysosomes showed LMP, their structure grossly remained after the execution of apoptosis. In contrast, the addition of 4-HNE caused “bursting” cell death with all lysosomes fading away prior to the vanishment of the cytoplasm and nuclear chromatin via necrosis. 4-HNE treatment of HepG2 cells caused a rapid vanishment of lysosomes via the explosive shrinkage in the early phase of cell degeneration, as clearly shown in Video (2.39 MB). 4-HNE caused a distinct disruption of the lysosomal limiting membranes (LMR) with the rapid and massive leakage of LysoTracker into the cytoplasm [32]. Consequently, all lysosomes disappeared in the early phase, prior to the execution of necrosis (Figure 5, upper column, circle). Most importantly, the intensity of lysosomal staining is not proportional to the extent of the lysosomal membrane disintegrity. In addition, tracing lysosomes until the execution of necrosis was impossible, whereas lysosomes could be grossly detected after the execution of apoptosis.
Figure 5.
Time-lapse imagings showing LMR (upper) and LMP (lower) and. LysoTracker is a highly soluble small molecule that is retained within the acidic lysosomal compartments in normal conditions, but shows an extralysosomal leakage at pathological states. HepG2 cells, the hepatocellular carcinoma cell line, were exposed to the synthetic 4-hydroxy-2-nonenal (4-HNE) or epirubicin (EPI). Inserts on each right top indicates morphological changes that were observed on the bright field imaging. The addition of 4-HNE resulted in the gradual reduction and loss of lysosomes prior to the occurrence of necrotic cell death (upper inserts). In contrast, lysosomes remained almost intact until the apoptotic cell death of EPI-treated cells (lower inserts). This indicates that necrosis occurs via LMR, whereas apoptosis occurs via LMP. Blue, Hoechst in the nucleus. Orange, LysoTracker retained within and around lysosomes. (Cited from [32]).
Figure 5.
Time-lapse imagings showing LMR (upper) and LMP (lower) and. LysoTracker is a highly soluble small molecule that is retained within the acidic lysosomal compartments in normal conditions, but shows an extralysosomal leakage at pathological states. HepG2 cells, the hepatocellular carcinoma cell line, were exposed to the synthetic 4-hydroxy-2-nonenal (4-HNE) or epirubicin (EPI). Inserts on each right top indicates morphological changes that were observed on the bright field imaging. The addition of 4-HNE resulted in the gradual reduction and loss of lysosomes prior to the occurrence of necrotic cell death (upper inserts). In contrast, lysosomes remained almost intact until the apoptotic cell death of EPI-treated cells (lower inserts). This indicates that necrosis occurs via LMR, whereas apoptosis occurs via LMP. Blue, Hoechst in the nucleus. Orange, LysoTracker retained within and around lysosomes. (Cited from [32]).
3. Lysosomal Membrane Permeabilization (LMP) and Apoptosis
As mentioned above, smaller perforations of the lysosomal limiting membrane cause a selective release of hydrolytic enzymes, whereas larger perforations cause a massive release of hydrolases [Figure 1B, light-orange]. For example, smaller dextran molecules (MW, 10 and 40 kDa) are released from the lysosome via LMP, whereas larger dextran molecules (70 and 250 kDa) are retained [33]. Further, cationic nanoparticles first induce the release of smaller cleaved-cathepsin D (~27 kDa), followed by the larger cathepsin B (~37 kDa) [7]. Nevertheless, the upper limit of size-selection alone does not always apply in all cell death models, because leakage of the bigger lysosomal protein like N-acetyl-b-glucosaminidase (150 kDa) was observed under certain experimental conditions [34–36]. Both the extent of LMP and the subset of lysosomes affected by LMR probably direct the downstream cellular responses to determine the cell fate; survival or death, or apoptosis or necrosis (Figure 1). In this sense, electron microscopic analysis of the lysosomal limiting membrane is indispensable to implicate LMR in the given cell death model of each experimental paradigms. As smaller perforations are hardly detected, LMP could not be detected by electron microscopy. One of the highly-sensitive methodology for detecting LMP would be the specific cytochemical procedure, for example, the lysosomal galectin puncta assay, which detects translocated galectins 1 and 3 much earlier than methods that monitor cathepsin release (Figure 1) [37]. The LysoTracker can also help distinguish the subset of damaged lysosomes from other intact lysosomes, as shown below.
Cathepsins are considered downstream mediators of the lysosomal cell death, but they can apparently initiate LMP. The promoting effect of LMP by cathepsins might be due to the intralysosomal degradation of highly-glycosylated lysosome-associated membrane proteins, i.e., the protective glycocalyx shield [38,39]. Further, minor leakage of cathepsins activates LMP by cleaving sphingosine kinase 1 that maintains lysosomal membrane stability [40,41]. LMP is induced also by phospholipase A2, which destabilizes purified lysosomes by degrading membrane phospholipids [42,43]. LMP is triggered by a wide range of apoptotic stimuli such as death receptor activation, radiation, cytotoxic drugs, viral and bacterial proteins, oxidative stress, endoplasmic reticulum stress, proteasome inhibition, DNA damage, osmotic stress, and growth factor starvation [22,44–47]. LMP activates apoptotic signaling and the intrinsic apoptosis pathway in the apoptosis-competent cells [14].
A key event in the execution of apoptosis is the release of apoptogenic factors from mitochondria. In 1998, Roberg and Öllinger discovered that a small amount of cathepsin D was released from lysosomes into the cytosol upon oxidative stress-induced apoptotic cell death [48]. Thereafter, the role of cathepsins as an executor of LMP-induced apoptosis was shown by the ability of microinjected cathepsin B or D to trigger mitochondrial outer membrane permeabilization (MOMP) [49,50]. LMP-induced apoptosis is usually activated via MOMP, either by activating cathepsin-mediated cleavage of pro-apoptotic proteins (e.g., Bax, Bak, Bid, Bad, Bim, Noxa, and Puma), or by inhibiting cleavage of anti-apoptotic Bcl-2 homologue proteins (e.g., Bcl-2, Bcl-XL and Mcl-1) [51–53]. Bax and Bak are pore-forming proteins that enable the release of apoptogenic factors such as cytochrome c from mitochondria. Cathepsins induce the proteolytic activation of substrates such as Bid and Bax, which in turn promotes MOMP and caspase activation [54,55]. However, since cathepsin release was observed in Bax/Bak-double-knockout cells, additional mechanisms independent of Bax and Bak may contribute to LMP [56]. Interestingly, amyloid β shows structural similarities to pore-forming bacterial toxins, and triggers LMP by creating pores [57]. During apoptosis, intracellular and extracellular perturbations converge in the MOMP-integration phase, resulting in the activation of caspases that serve as the final executors and dismantle cell components and nuclear DNA.
4. Lysosomal Membrane Rupture (LMR) and Necrosis
Unlike mitochondria, lysosomes lack antioxidant enzymes such as superoxide dismutase, catalase, or glutathione peroxidase. Therefore, when ROS levels are high in the cell, ROS can readily cross and damage lysosomal limiting membranes. Intralysosomal iron (Fe2+), generated from iron-containing metalloproteins, reacts with hydrogen peroxide (H2O2) in lysosomes. This generates Fe3+ and highly reactive hydroxyl radicals (HO) to cause lipid peroxidation of the lysosomal membrane [59–61]. For example, macrophages that ingested silica particles with abundant surface-bound iron suffered from extensive lysosomal damage, whereas those that ingested silica particles pretreated with the pharmacologic iron chelator displayed only modest lysosomal leakage [62]. Phagocytosis of iron complexes or iron-containing proteins increases lysosomal vulnerability, whereas a reduction in the intralysosomal reactive iron reduces lysosomal leakage and cell death [63,64]. These data altogether indicate that iron is indispensable for inducing lysosomal membrane disintegrity. 4-HNE is derived not only from the dietary linoleic acids but also from the cardiolipin of mitochondrial inner membranes. Hydroxyl radicals induce peroxidation of cardiolipin and thereby generate endogenous 4-HNE. Since abundant ROS are produced during β-oxidation of the palmitate in mitochondria, an increased amount of 4-HNE is endogenously generated from linoleic acids which are involved in cardiolipin [4]. 4-HNE being generated from both dietary and mitochondrial sources, forms adducts with different side chains in proteins, namely Cys, His, Lys, and Arg. In a rat model with oxidative stress, Carbone et al. showed by mass spectral analysis that Hsp72 treated with 4-HNE caused adduct formation at Cys267 in the ATPase domain [65]. In addition, 4-HNE-adducted Hsp70 has been identified in the G93A-SOD1 transgenic mice, a model of familial amyotrophic lateral sclerosis [66]. It is likely that modifications of Hsp72 or 70 (also called Hsp70.1 in humans) is closely related to diseases.
In 1962, the heat shock protein (Hsp) was discovered in Drosophila melanogaster after the heat shock [67]. Later, it was demonstrated that other than thermal stress, Hsp expression is induced by such insults as ischemia, irradiation, infection, inflammation, and exposure to organics and oxidants [68]. Hsp70.1 is responsible for folding not only newly-synthesized polypeptides under physiological conditions but also misfolded proteins under cellular stresses. Hsp70.1 recruits damaged/aged proteins to the proteasome for turnover [69]. Further, Hsp70.1 transports damaged/aged proteins to lysosomes for recycling amino acids via chaperone-mediated autophagy [70], and, most importantly, contributes to preserving the lysosomal membrane integrity. Hsp70.1 exerts dual roles as a chaperone protein for the transport of garbage proteins and as a stabilizer for the lysosomal limiting membrane. The latter function is achieved by both sphingolipid composition and acid sphingomyelinase (EC3.1.4.12) [71]. Acid sphingomyelinase resides inside lysosomal lumen and its hydrolytic activity is stabilized by bis(monoacylglycero)phosphate (BMP) [72]. The Hsp70.1-BMP interaction enhances association of BMP with acid sphingomyelinase. This can activate this enzyme, and breakdown sphingomyelin to generate ceramide which protects the lysosomal membrane from rupturing [25,26,73,74]. Accordingly, disorders of Hsp70.1 lead not only to autophagy failure but also to lysosomal disintegrity.
The hippocampal CA1 tissue on days 3 and 5 after transient brain ischemia showed a remarkable (3-9 fold) upregulation of Hsp70.1 on the 2-D oxyblot analysis (Figure 6A). Further, the Matrix-assisted laser desorption ionization-time of flight/time of flight analysis showed a decrease of its molecular weight from 157.20 to 113.12. This means that the specific oxidative injury ‘carbonylation’ occurred at the Arg469 of Hsp70.1 due to ROS being generated during the reperfusion phase (Figure 6B,C) [30,31]. In addition, using various brain tissues of non-ischemic monkeys, the calpain-mediated cleavage of the carbonylated Hsp70.1 was demonstrated in vitro (Figure 6D) [75,76]. Calpain alone without 4-HNE treatment (Figure 6D, time point (‘0’) showed no cleavage of Hsp70.1, but calpain-mediated Hsp70.1 cleavage progressed time-dependantly after incubation with 4-HNE (Figure 6D). Therefore, 4-HNE-induced carbonylation was thought indispensable to facilitate calpain-mediated cleavage of Hsp70.1 [77].
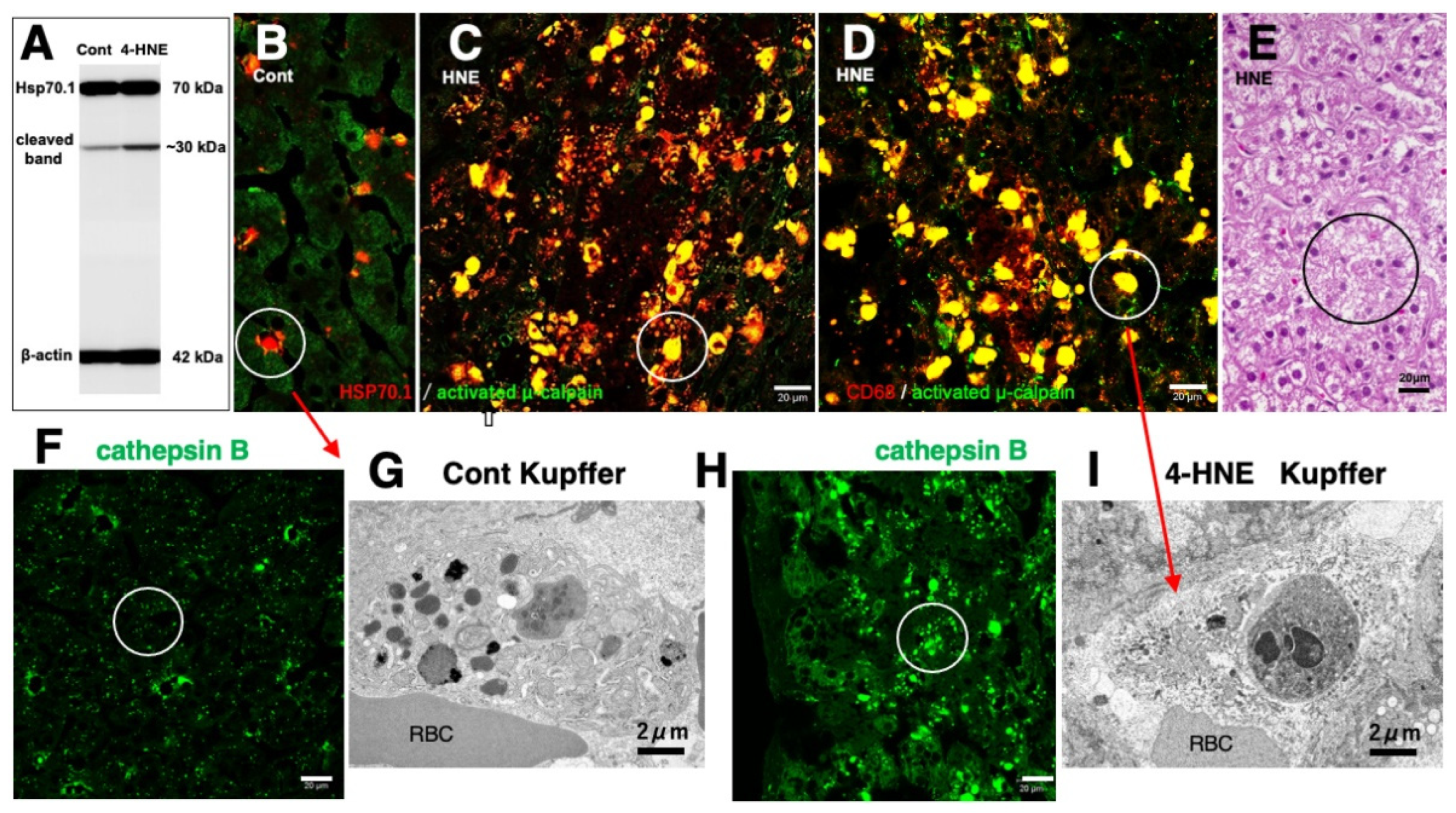
Similar to the in-vitro experiments using the hippocampal tissues (Figure 6D), the Western blotting analysis of the liver of monkeys which were injected 4-HNE for 24 weeks (total amount 120 mg) also indicates that the calpain-mediated cleavage of Hsp70.1 was increased by 4-HNE in the living animals (Figure 7A) [78]. By the immunofluorescence analysis of the control liver, activated μ-calpain was observed in the Kupffer cells in the vicinity of sinusoids, whereas hepatocytes showed negligible immunoreactivity (Figure 7B). In contrast, after the 4-HNE injections, the hepatocytes showed a remarkable increase in the merged immunoreactivity of Hsp70.1 and activated μ-calpain (Figure 7C, yellow). Especially, CD68-positive Kupffer cells showed an extensive merged immunoreactivity of Hsp70.1 and activated μ-calpain (Figure 7C,D, circles). Perisinusoidal hepatocytes showed necrosis with a complete dissolution of cytoplasmic organelle (Figure 7E, circle), which was consistent with necrosis of CA1 neurons after transient ischemia [8]. Morphological evidence of apoptosis such as apoptotic bodies and nuclear blebbings was not observed in both neurons after transient ischemia and hepatocytes after the 4-HNE treatment. These in-vitro and in-vivo results suggested that activated μ-calpain interacted with Hsp70.1 after 4-HNE injections, which conceivably facilitated the calpain-mediated cleavage of carbonylated Hsp70.1. In parallel with hepatocyte degeneration/necrosis, cathepsin B immunoreactivity showed an increase in the cytoplasm of both hepatocytes and Kupffer cells (Figure 7H), compared to the control (Figure 7F). The disintegration of lysosomes was much stronger in the Kupffer cells, compared to the hepatocytes. Most of the lysosomes in the Kupffer cells faded away due to LMR (Figure 7G,I).
Figure 6.
Upregulation, carbonylation, and cleavage of Hsp70.1 in the monkey hippocampal CA1 tissues after transient global brain ischemia. Hsp70.1 carbonylation is upregulated on days 3 and 5 after ischemia (A), and carbonylation occurs at the key site Arg469 (R*) of Hsp70.1 (B,C) via the proteomics analysis. Calpain-mediated cleavage of carbonylated Hsp70.1 in vitro increases time-dependantly after the 4-HNE treatment (D). Since Hsp70.1 exerts dual roles as a chaperone protein and a lysosomal stabilizer, its disorder causes cell degeneration/death via the lysosomal membrane disintegrity. (Reprinted from [75]).
Figure 6.
Upregulation, carbonylation, and cleavage of Hsp70.1 in the monkey hippocampal CA1 tissues after transient global brain ischemia. Hsp70.1 carbonylation is upregulated on days 3 and 5 after ischemia (A), and carbonylation occurs at the key site Arg469 (R*) of Hsp70.1 (B,C) via the proteomics analysis. Calpain-mediated cleavage of carbonylated Hsp70.1 in vitro increases time-dependantly after the 4-HNE treatment (D). Since Hsp70.1 exerts dual roles as a chaperone protein and a lysosomal stabilizer, its disorder causes cell degeneration/death via the lysosomal membrane disintegrity. (Reprinted from [75]).
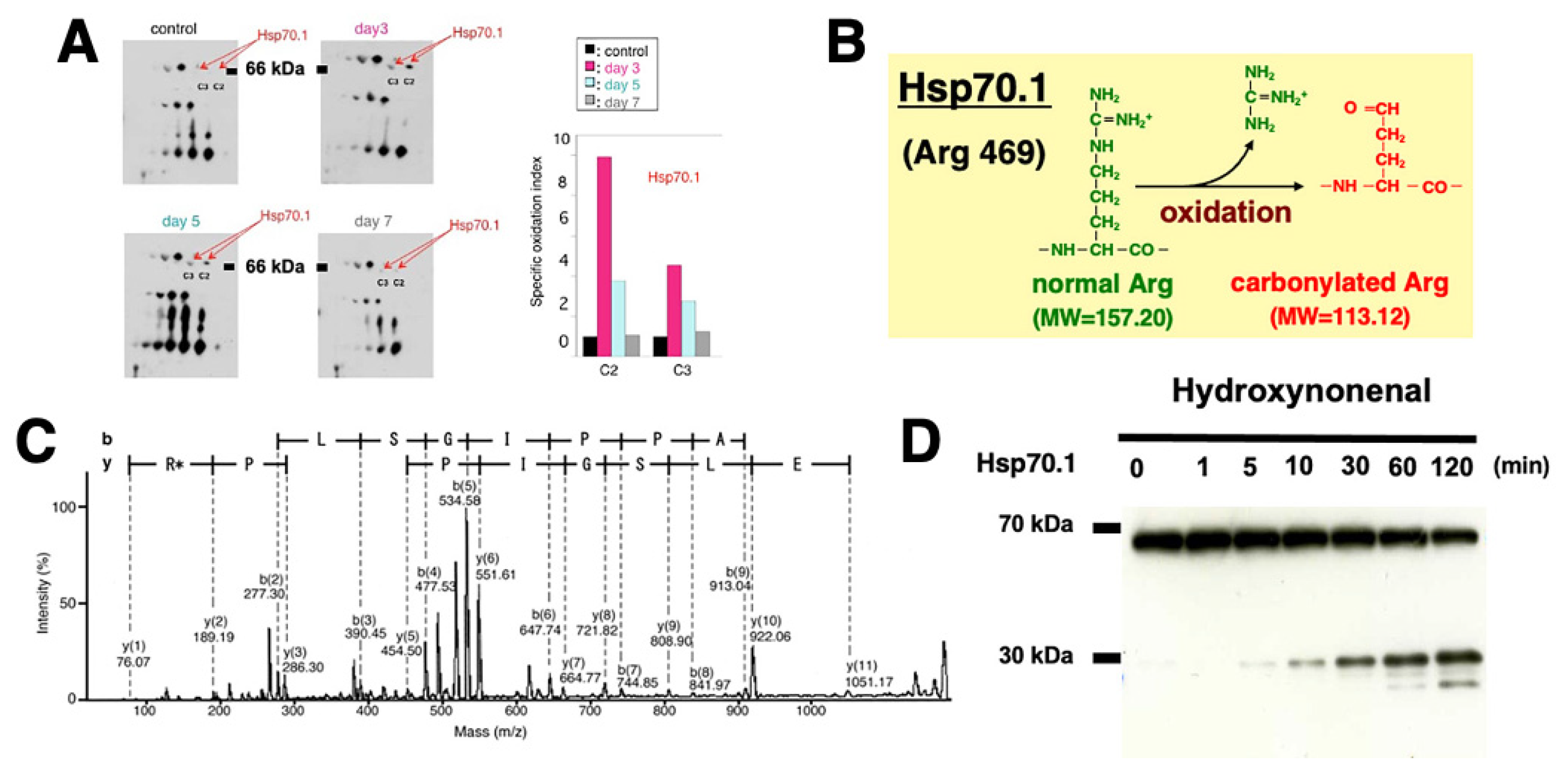
Figure 7.
Possible interaction of activated μ-calpain with carbonylated Hsp70.1 in the monkey liver after the intravenous 4-HNE injections (5mg/w x 24 weeks). On Western blotting, calpain-mediated cleavage of Hsp70.1 increases in the liver tissue after the 4-HNE treatment (A). By immunofluorescence histochemistry, merged immunoreactivity (yellow) of activated μ-calpain (green) and Hsp70.1 (red) is remarkably increased not only in hepatocytes (C, circle) but also in CD68-positive Kupffer cells (D, circle) after the 4-HNE treatment, as compared to the control (B). Lysosomal loss occurs in the perisinusoidal Kupffer cells after the 4-HNE treatment (G,I). The extralysosomal release of cathepsin B (F,H, circles) in hepatocytes caused their necrotic cell death (E, circle). Apoptotic bodies were not seen in both Kupffer cells and hepatocytes. RBC, red blood cell within the sinusoid. (Cited and adapted from [26,78]).
Figure 7.
Possible interaction of activated μ-calpain with carbonylated Hsp70.1 in the monkey liver after the intravenous 4-HNE injections (5mg/w x 24 weeks). On Western blotting, calpain-mediated cleavage of Hsp70.1 increases in the liver tissue after the 4-HNE treatment (A). By immunofluorescence histochemistry, merged immunoreactivity (yellow) of activated μ-calpain (green) and Hsp70.1 (red) is remarkably increased not only in hepatocytes (C, circle) but also in CD68-positive Kupffer cells (D, circle) after the 4-HNE treatment, as compared to the control (B). Lysosomal loss occurs in the perisinusoidal Kupffer cells after the 4-HNE treatment (G,I). The extralysosomal release of cathepsin B (F,H, circles) in hepatocytes caused their necrotic cell death (E, circle). Apoptotic bodies were not seen in both Kupffer cells and hepatocytes. RBC, red blood cell within the sinusoid. (Cited and adapted from [26,78]).
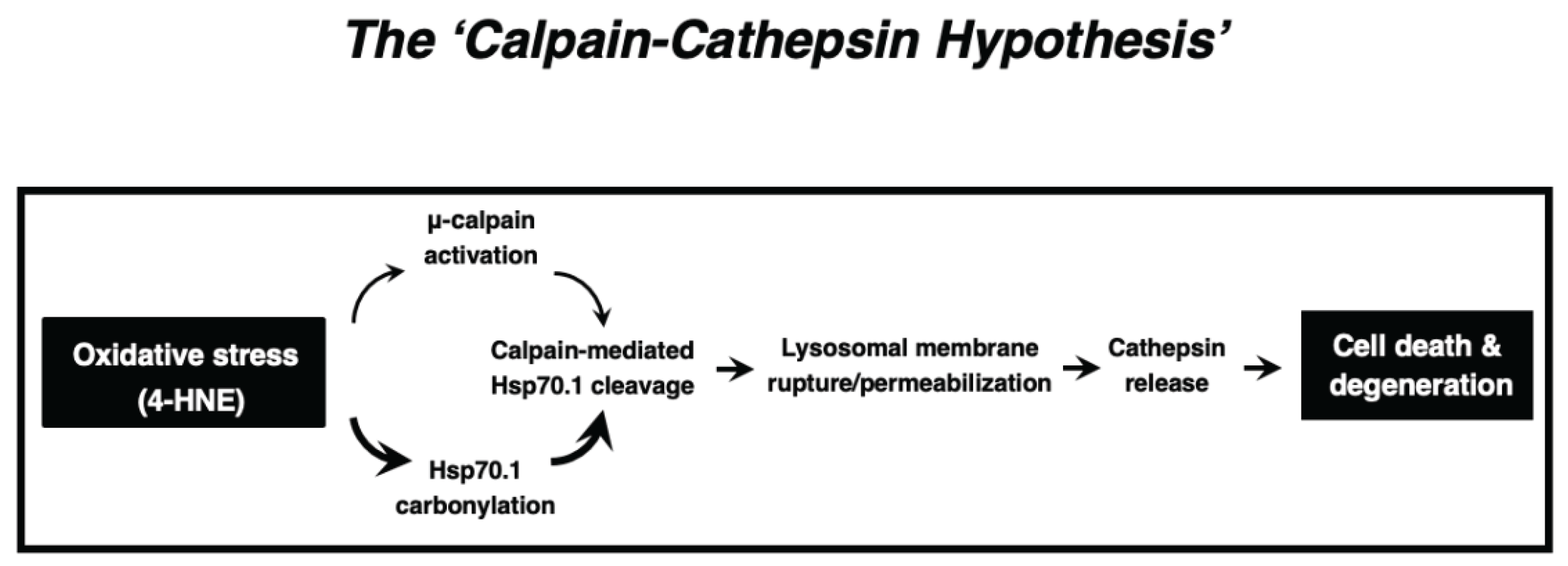
In summary, the available in-vitro and in-vivo data which were obtained from C. elegans to humans, and from neurons to hepatocytes, combined together, indicate that ‘calpain-mediated cleavage of the carbonylated Hsp70.1’ induced cell degeneration/death via LMR which was associated with the massive release of cathepsin enzymes. The calpain-cathepsin hypothesis (Figure 8) which was formulated by Yamashima et al. in 1998 [23] and modified in 2009 [31], can explain the mechanism of necrotic cell death of neurons, hepatocytes, and Kupffer cells. Importantly, the ultrastructural evidence of LMR was found not after cell death or disease is completed, but during the progression phase of cell degeneration.
5. Conclusions
LMP can be detected by cytochemistry but not by electron microscopy, because of the difficulty of detecting tiny pores at the lysosomal limiting membrane. Since LMR may occur within minutes or hours being associated with lysosomal fading, we have a very restricted chance of finding very small numbers of LMR in the degenerating/dying cells at the given time point. Careful and detailed electron microscopic analyses are indispensable to detect LMR.
Funding
This work was supported by a grant from Kiban-Kenkyu (B) (19H04029) from the Japanese Ministry of Education, Culture, Sports, Science and Technology.
Institutional Review Board Statement
The protocol of monkey experiments conducted by the author’s group (Figures 2C, 3, 6, 7 and 8) was approved by the Committee on the Ethics of Animal Experiments of Kanazawa University (Protocol Number: AP-194062, AP24-046).
Acknowledgments
The author is deeply grateful for many colleagues participating in the monkey experiments from 1990 to 2025.
Conflicts of Interest
The author declares no conflicts of interest.
Abbreviations
| BMP | bis(monoacylglycero)phosphate |
| CA1 | cornu Ammonis 1 |
| CLEAR | coordinated lysosomal expression and regulation |
| EPI | epirubicin |
| ESCRT | endosomal sorting complex required for transport |
| 4-HNE | 4-hydroxy-2-nonenal |
| Hsp | heat-shock protein |
| Hsp70.1 | heat-shock protein 70.1 |
| IL-1β | interleukin-1β |
| LAMP-2 | lysosome-associated membrane protein 2 |
| LIMP-2 | lysosomal integral membrane protein type 2 |
| LMP | lysosomal membrane permeabilization |
| LMR | lysosomal membrane rupture |
| MOMP | mitochondrial outer membrane permeabilization |
| PUFA | polyunsaturated fatty acids |
| TFEB | transcription factor EB |
References
- De Duve, C.; Pressman, B.C., Gianetto, R.; Wattiaux, R.; Appelmans, F. Tissue fractionation studies. 6. Intracellular distribution patterns of enzymes in rat-liver tissue. Biochem. J. 1955, 60, 604–617. [CrossRef]
- Luzio, J.P.; Pryor, P.R.; Bright, N.A. Lysosomes: fusion and function. Nat. Rev. Mol. Cell Biol. 2007, 8, 622-632. [CrossRef]
- Saftig, P.; Klumperman, J. Lysosome biogenesis and lysosomal membrane proteins: trafficking meets function. Nat. Rev. Mol. Cell Biol. 2009, 10, 623-635. [CrossRef]
- Yamashima, T. 4-Hydroxynonenal from mitochondrial and dietary sources causes lysosomal cell death for lifestyle-related diseases. Nutrients 2024, 16, 4171. [CrossRef]
- Sardiello, M.; Palmieri, M.; di Ronza, A.; Medina, D.L.; Valenza, M.; Gennarino, V.A.; Di Malta, C.; Donaudy, F.; Embrione, V.; Polishchuk, R.S. et al. A gene network regulating lysosomal biogenesis and function. Science 2009, 325, 473-477. [CrossRef]
- Skowyra, M.L.; Schlesinger, P.H.; Naismith, T.V.; Hanson, P.I. Triggered recruitment of ESCRT machinery promotes endolysosomal repair. Science 2018; 360, 6384. [CrossRef]
- Wang F, Gómez-Sintes R, Boya P. Lysosomal membrane permeabilization and cell death. Traffic 2018, 19, 918-931. [CrossRef]
- Yamashima, T.; Saido, T.C.; Takita, M.; Miyazawa, A.; Yamano, J.; Miyakawa, A.; Nishijyo, H.; Yamashita, J.; Kawashima, S.; Ono, T.; et al. Transient brain ischaemia provokes Ca2+, PIP2 and calpain responses prior to delayed neuronal death in monkeys. Eur. J. Neurosci. 1996, 8, 1932–1944. [CrossRef]
- de Duve, C.; Wattiaux, R. Functions of lysosomes. Ann. Rev. Physiol. 1966, 28, 435–492. [CrossRef]
- Brunk, U.T.; Ericsson, J.L. Cytochemical evidence for the leakage of acid phosphatase through ultrastructurally intact lysosomal membranes. Histochem. J. 1972, 4, 479–491. [CrossRef]
- Brunk, U.T.; Svensson, I. Oxidative stress, growth factor starvation and Fas activation may all cause apoptosis through lysosomal leak. Redox Rep. 1999, 4, 3–11. [CrossRef]
- Li, W.; Yuan, X.; Nordgren, G.; Dalen, H.; Dubowchik, G.M.; Firestone, R.A.; Brunk, U.T. Induction of cell death by the lysosomotropic detergent MSDH. FEBS Lett. 2000, 470, 35–39. [CrossRef]
- Antunes, F.; Cadenas, E.; Brunk, U.T. Apoptosis induced by exposure to a low steady-state concentration of H2O2 is a consequence of lysosomal rupture. Biochem. J. 2001, 356 Pt 2, 549–555. [CrossRef]
- Kågedal, K.; Zhao, M.; Svensson, I.; Brunk, U.T. Sphingosine-induced apoptosis is dependent on lysosomal proteases. Biochem. J. 2001, 359, 335–343. [CrossRef]
- Yamashima, T.; Seike, T.; Oikawa, S.; Kobayashi, H.; Kido, H.; Yanagi, M.; Yamamiya, D.; Li, S.; Boontem, P.; Mizukoshi, E. Hsp70.1 carbonylation induces lysosomal cell death for lifestyle-related diseases. Front. Mol. Biosci. 2023. 9, 1063632. [CrossRef]
- Terman, A.; Kurz, T.; Gustafsson, B.; Brunk, U.T. Lysosomal labilization. IUBMB Life 2006, 58, 531–539. [CrossRef]
- Stern, S.T., Adiseshaiah, P.P.; Crist, R.M. Autophagy and lysosomal dysfunction as emerging mechanisms of nanomaterial toxicity. Part Fibre Toxicol 2012, 9, 20. [CrossRef]
- Li, Y.; Chen, B.; Zou,W.; Wang, X.; Wu, Y.; Zhao, D.; Sun, Y.; Liu, Y.; Chen, L.; Miao, L.; et al. The lysosomal membrane protein SCAV-3 maintains lysosome integrity and adult longevity. J. Cell Biol. 2016, 215, 167–185.
- Yamashima, T. Reconsider Alzheimer’s disease by the ‘calpain-cathepsin hypothesis’—A perspective review. Prog. Neurobiol. 2013, 105, 1–23. [CrossRef]
- Bursch,W. The autophagosomal-lysosomal compartment in programmed cell death. Cell Death Differ. 2001, 8, 569–581. [CrossRef]
- Nylandsted, J.; Gyrd-Hansen, M.; Danielewicz, A.; Fehrenbacher, N.; Lademann, U.; Høyer-Hansen, M.;Weber, E.; Multhoff, G.; Rohde, M.; Jäättelä, M. Heat shock protein 70 promotes cell survival by inhibiting lysosomal membrane permeabilization. J. Exp. Med. 2004, 200, 425–435. [CrossRef]
- Kirkegaard, T.; Jäättelä, M. Lysosomal involvement in cell death and cancer. Biochim. Biophys. Acta 2009, 1793, 746–754. [CrossRef]
- Yamashima, T.; Kohda, Y.; Tsuchiya, K.; Ueno, T.; Yamashita, J.; Yoshioka, T.; Kominami, E. Inhibition of ischaemic hippocampal neuronal death in primates with catepsin B inhibitor CA-074: A novel strategy for neuroprotection based on ‘calpain-cathepsin hypothesis’. Eur. J. Neurosci. 1998, 10, 1723–1733. [CrossRef]
- Yamashima, T.; Ota, T.; Mizukoshi, E.; Nakamura, H.; Yamamoto, Y.; Kikuchi, M.; Yamashita, T.; Kaneko, S. Intake of ω-6 polyunsaturated fatty acid-rich vegetable oils and risk of lifestyle diseases. Adv. Nutr. 2020, 11, 1489–1509. [CrossRef]
- Yamashima, T. Implication of cysteine proteases calpain, cathepsin and caspase in ischemic neuronal death of primates. Prog. Neurobiol. 2000, 62, 273–295. [CrossRef]
- Yamashima, T. Implication of vegetable oil-derived hydroxynonenal in the lysosomal cell death for lifestyle-related diseases. Nutrients 2023, 15, 609. [CrossRef]
- Syntichaki, P.; Xu, K.; Driscoll, M.; Tavernarakis, N. Specific aspartyl and calpain proteases are required for neurodegeneration in C. elegans. Nature 2002, 419, 939–944. [CrossRef]
- Tardy, C.; Codogno, P.; Autefage, H.; Levade, T.; Andrieu-Abadie, N. Lysosomes and lysosomal proteins in cancer cell death (new players of an old struggle). Biochim. Biophys. Acta 2006, 1765, 101–125. [CrossRef]
- Brunk, U.T.; Dalen, H.; Roberg, K.; Hellquist, H.B. Photo-oxidative disruption of lysosomal membranes causes apoptosis of cultured human fibroblasts. Free Rad. Biol. Med. 1997, 23, 616–626. [CrossRef]
- Oikawa, S., Yamada, T., Minohata, T., Kobayashi, H., Furukawa, A., Tada-Oikawa, S., et al. Proteomic identification of carbonylated proteins in the monkey hippocampus after ischemia-reperfusion. Free Radic. Biol. Med. 2009. 46, 1472–1477. [CrossRef]
- Yamashima, T., and Oikawa, S. The role of lysosomal rupture in neuronal death. Prog. Neurobiol. 2009. 89, 343–358. [CrossRef]
- Seike, T.; Boontem, P.; Kido, H.; Yanagi, M.; Yamamiya, D.; Nakagawa, H.; Yamashita, T.; Li, S.; Okada, H.; Harada, K.; et al. Hydroxynonenal causes hepatocyte death by disrupting lysosomal integrity in non-alcoholic steatohepatitis. Cell. Mol. Gastro. Hepatol. 2022, 14, 925–944. [CrossRef]
- Bidère N, Lorenzo HK, Carmona S, et al. Cathepsin D triggers Bax activation, resulting in selective apoptosis-inducing factor (AIF) relocation in T lymphocytes entering the early commitment phase to apoptosis. J Biol Chem. 2003; 278(33): 31401-31411. [CrossRef]
- Ono, K.; Kim, S.O.; Han, J. Susceptibility of lysosomes to rupture is a determinant for plasma membrane disruption in tumor necrosis factor alpha-induced cell death. Mol. Cell Biol. 2003. 23, 665–676. [CrossRef]
- Kågedal, K.; Johansson, A.C.; Johansson, U.; Heimlich, G.; Roberg, K.; Wang, N.S.; Jürgensmeier, J.M.; Öllinger, K. Lysosomal membrane permeabilization during apoptosis-involvement of Bax? Int. J. Exp. Pathol. 2005, 86, 309–321. [CrossRef]
- Blomgran, R.; Zheng, L.; Stendahl, O. Cathepsin-cleaved Bid promotes apoptosis in human neutrophils via oxidative stress-induced lysosomal membrane permeabilization. J. Leukoc. Biol. 2007. 81, 1213–1223. [CrossRef]
- Aits S, Kricker J, Liu B, Ellegaard AM, Hämälistö S, Tvingsholm S, Corcelle-Termeau E, Høgh S, Farkas T, Holm Jonassen A, et al. Sensitive detection of lysosomal membrane permeabilization by lysosomal galectin puncta assay. Autophagy 2015, 11, 1408-1424. [CrossRef]
- Eskelinen, E.L.; Tanaka, Y.; Saftig, P. At the acidic edge: emerging functions for lysosomal membrane proteins. Trends Cell Biol. 2003, 13, 137-145. [CrossRef]
- Fehrenbacher, N.; Bastholm, L.; Kirkegaard-Sørensen, T.; Rafn, B., Bøttzauw, T.; Nielsen, C.; Weber, E.; Shirasawa, S.; Kallunki, T.; Jäättelä M. Sensitization to the lysosomal cell death pathway by oncogene-induced down-regulation of lysosome-associated membrane proteins 1 and 2. Cancer Res. 2008. 68, 6623-6633. [CrossRef]
- Mora, R.; Dokic, I.; Kees, T.; Hüber, C.M.; Keitel, D.; Geibig, R.; Brügge, B.; Zentgraf, H.; Brady, N.R; Régnier-Vigouroux, A. Sphingolipid rheostat alterations related to transformation can be exploited for specific induction of lysosomal cell death in murine and human glioma. Glia 2010, 58, 1364-1383. [CrossRef]
- Taha, T.A.; Kitatani, K.; Bielawski, J.; Cho, W.; Hannun, Y. A.; Obeid, L.M. Tumor necrosis factor induces the loss of sphingosine kinase-1 by a cathepsin B-dependent mechanism. J. Biol. Chem. 2005, 280, 17196-17202. [CrossRef]
- Zhao, M.; Brunk, U.T.; Eaton, J.W. Delayed oxidant-induced cell death involves activation of phospholipase A2. FEBS Lett. 2001. 509, 399-404.
- Zhao, M.; Antunes, F.; Eaton, J.W.; Brunk, U.T. Lysosomal enzymes promote mitochondrial oxidant production, cytochrome c release and apoptosis. Eur. J. Biochem. 2003, 270, 3778-3786. [CrossRef]
- Boya, P.; Kroemer, G. Lysosomal membrane permeabilization in cell death. Oncogene 2008, 27, 6434–6451. [CrossRef]
- Guicciardi, M.E.; Leist, M.; Gores, G.J. Lysosomes in cell death. Oncogene 2004, 23, 2881–2890. [CrossRef]
- Chwieralski, C.E.; Welte, T.; Bühling F. Cathepsin-regulated apoptosis. Apoptosis 2006, 11, 143–149. [CrossRef]
- Stoka, V.; Turk, V.; Turk, B. Lysosomal cysteine cathepsins: signaling pathways in apoptosis. Biol. Chem. 2007, 388, 555–560. [CrossRef]
- Roberg, K.; Öllinger, K. Oxidative stress causes relocation of the lysosomal enzyme cathepsin D with ensuing apoptosis in neonatal rat cardiomyocytes. Am. J. Pathol. 1998, 152, 1151-1156. PMID: 9588882; PMCID: PMC1858594.
- Bivik, C.A.; Larsson, P.K.; Kågedal, K.M.; Rosdahl, I.K.; Öllinger, K.M. UVA/B-induced apoptosis in human melanocytes involves translocation of cathepsins and Bcl-2 family members. J. Invest. Dermatol. 2006, 126, 1119-1127. [CrossRef]
- Roberg, K., Kågedal, K. and Öllinger, K. Microinjection of cathepsin D induces caspase-dependent apoptosis in fibroblasts. Am. J. Pathol. 2002, 161, 89-96. [CrossRef]
- Appelqvist, H.; Johansson, A. C.; Linderoth, E.; Johansson, U.; Antonsson, B.; Steinfeld, R.; Kågedal, K.; Öllinger, K. Lysosome-mediated apoptosis is associated with cathepsin D-specific processing of bid at Phe24, Trp48, and Phe183. Ann. Clin. Lab. Sci. 2012, 42, 231-242. https://www.annclinlabsci.org/content/42/3/231.full.pdf+html.
- Cirman, T.; Oresić, K.; Mazovec, G.D.; Turk, V.;Reed, J.C.; Myers, R.M.; Salvesen, G.S.; Turk, B. Selective disruption of lysosomes in HeLa cells triggers apoptosis mediated by cleavage of Bid by multiple papain-like lysosomal cathepsins. J. Biol. Chem. 2004, 279, 3578-3587. [CrossRef]
- Droga-Mazovec, G.; Bojic, L.; Petelin, A.; Ivanova, S.; Romih, R.; Repnik, U.; Salvesen, G. S.; Stoka, V.; Turk, V.; Turk, B. Cysteine cathepsins trigger caspase-dependent cell death through cleavage of bid and antiapoptotic Bcl-2 homologues. J. Biol. Chem. 2008, 283, 19140-19150. [CrossRef]
- Aits S, and Jäättelä, M. Lysosomal cell death at a glance. J. Cell Sci. 2013, 126, 1905-1912. [CrossRef]
- Wang, F.; Salvati, A.; Boya, P. Lysosome-dependent cell death and deregulated autophagy induced by amine-modified polystyrene nanoparticles. Open Biol. 2018, 8, 170271. [CrossRef]
- Boya, P.; Andreau, K.; Poncet ,D.; Zamzami, N.;, Perfettini, J.L.; Metivier, D.; Ojcius, D.M.; Jäättelä, M.; Kroemer, G. Lysosomal membrane permeabilization induces cell death in a mitochondrion-dependent fashion. J. Exp. Med. 2003 197, 1323-1334. [CrossRef]
- Johansson, A.C.; Appelqvist, H.; Nilsson, C.; Kågedal, K.; Roberg, K.; Öllinger, K. Regulation of apoptosis-associated lysosomal membrane permeabilization. Apoptosis. 2010 15, 527-40. [CrossRef]
- Eaton, J.W.; Qian, M. Molecular bases of cellular iron toxicity. Free Radic. Biol. Med. 2002; 32, 833-840. [CrossRef]
- Kurz, T.; Gustafsson, B.; Brunk, U.T. Intralysosomal iron chelation protects against oxidative stress-induced cellular damage. FEBS J. 2006, 273, 3106-3117. [CrossRef]
- Krenn MA, Schurz M, Teufl B, Uchida K, Eckl PM, Bresgen N. Ferritin-stimulated lipid peroxidation, lysosomal leak, and macroautophagy promote lysosomal “metastability” in primary hepatocytes determining in vitro cell survival. Free Radic. Biol. Med. 2015, 80, 48-58. [CrossRef]
- Persson, H.L. Iron-dependent lysosomal destabilization initiates silica-induced apoptosis in murine macrophages. Toxicol. Lett. 2005, 159, 124–133. [CrossRef]
- Garner, B.; Li, W.; Roberg, K.; Brunk, U.T. On the cytoprotective role of ferritin in macrophages and its ability to enhance lysosomal stability. Free Radic. Res. 1997, 27, 487–500. [CrossRef]
- Yu, Z.; Persson, H.L.; Eaton, J.W.; Brunk, U.T. Intralysosomal iron: a major determinant of oxidant-induced cell death. Free Radic. Biol. Med. 2003, 34, 1243–1252.
- Carbone, D.L.; Doorn, J.A.; Kiebler, Z.; Sampey, B.P.; Petersen, D.R. Inhibition of Hsp72-mediated protein refolding by 4-hydroxy-2-nonenal. Chem. Res. Toxicol. 2004, 17, 1459–1467. [CrossRef]
- Perluigi, M.; Poon, H.F.; Hensley, K.; Pierce, W.M.; Klein, J.B.; Calabrese, V.; de Marco, C.; Butterfield, D.A. Proteomic analysis of 4-hydroxy-2-nonenal-modified proteins in G93A-SOD1 transgenic mice – a model of familial amyotrophic lateral sclerosis. Free Radic. Biol. Med. 2005, 38, 960–968. [CrossRef]
- Ritossa, F. A new puffing pattern induced by temperature shock and DNP in Drosophila. Experientia 1962, 18, 571–573. [CrossRef]
- Lindquist, S.; Craig, E.A. The heat-shock proteins. Annu. Rev. Genet. 1988, 22, 631–677. [CrossRef]
- Demand, J.; Luders, J.; Hohfeld, J. The carboxy-terminal domain of Hsc70 provides binding sites for a distinct set of chaperone cofactors. Mol. Cell Biol. 1998, 18, 2023–2028. [CrossRef]
- Majeski, A.E.; Dice, J.F. Mechanisms of chaperone-mediated autophagy. Int. J. Biochem. Cell Biol. 2004, 36, 2435–2444. [CrossRef]
- Gabande-Rodriguez, E.; Boya, P.; Labrador, V.; Dotti, C.G.; Ledesma, M.D. High sphingomyelin levels induce lysosomal damage and autophagy dysfunction in Niemann Pick disease type A. Cell Death Differ. 2014, 21, 864–875. [CrossRef]
- Linke, T.; Wilkening, G.; Lansmann, S.; Moczall, H.; Bartelsen, O.; Weisgerber, J.; Sandhoff, K. Stimulation of acid sphingomyelinase activity by lysosomal lipids and sphingolipid activator proteins. Biol. Chem. 2001, 382, 283–290. [CrossRef]
- Heinrich, M.; Wickel, M.; Winoto-Morbach, S.; Schneider-Brachert, W.; Weber, T.; Brunner, J., Saftig P, Peters C, Krönke M, Schütze S. Ceramide as an activator lipid of cathepsin D. Adv. Exp. Med. Biol. 2000, 477, 305–315. [CrossRef]
- Kirkegaard, T.; Roth, A.G.; Petersen, N.H.; Mahalka, A.K.; Olsen, O.D.; Moilanen, I., Zylicz A, Knudsen J, Sandhoff K, Arenz C, Kinnunen PK, Nylandsted J, Jäättelä M. Hsp70 stabilizes lysosomes and reverts Niemann-Pick disease-associated lysosomal pathology. Nature 2010, 463, 549–553. [CrossRef]
- Yamashima, T.; Mathivanan, A.; Dazortsava, M.Y.; Sakai, S.; Kurimoto, S.; Zhu, H.; Funaki, N.; Liang, H.; Hullin-Matsuda, F.; Kobayashi, T.; Akatsu, H.; Takahashi, H.; Minabe, Y. Calpain-mediated Hsp70.1 cleavage in monkey CA1 after ischemia induces similar ‘lysosomal vesiculosis’ to Alzheimer neurons. J. Alzheimers Dis. Park. 2014, 4, 2. [CrossRef]
- Yamashima, T.; Seike, T.; Mochly-Rosen, D.; Chen, C.H.; Kikuchi, M.; Mizukoshi, E. Implication of the cooking oil-peroxidation product “hydroxynonenal” for Alzheimer’s disease. Front. Aging Neurosci. 2023. 15, 1211141. [CrossRef]
- Liang, H.; Kurimoto, S.; Shima, K.R.; Shimizu, D.; Ota, T.; Minabe, Y.,Yamashima, T. Why is hippocampal CA1 especially vulnerable to ischemia? SOJ Biochem. 2016, 2, 1–7. [CrossRef]
- Sahara, S.; Yamashima, T. Calpain-mediated Hsp70.1 cleavage in hippocampal CA1 neuronal death. Biochem. Biophys. Res. Commun. 2010, 393, 806–811. [CrossRef]
- Yamashima, T.; Mori, Y.; Seike, T.; Ahmed, S.; Boontem, P.; Li, S.; Oikawa, S.; Kobayashi, H.; Yamashita, T.; Kikuchi, M.; et al. Vegetable oil-peroxidation product ‘hydroxynonenal’ causes hepatocyte injury and steatosis via Hsp70.1 and BHMT disorders in the monkey liver. Nutrients 2023, 15, 1904. [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



